2018 Clinical & Dermatopathologic Cases
1. Peripheral T-cell lymphoproliferative disorder, NOS CD4-/CD8-/BF-1+
2. Blastic Plasmacytoid Dendritic Cell Neoplasm- An Underrecognoized but Deadly tumor
3A. A Unique Case of Extranodal NK/T-Cell Lymphoma Mimicking Mycosis Fungoides
4A. Congenital Leukemia Cutis as Presenting Feature of Acute Myeloid Leukemia
4B. Leukemic Infiltrate Secondary to Chronic Myelogenous Leukemia
5B. Pigmented Spindle Cell (Reed’s) Nevus with Chromosomal Mutations
6. A Case of Desmoplastic Malignant Melanoma: A Diagnostic Pitfall
7. Ectopic Extramammary Paget’s Disease Presenting on the Face - An Extraordinary Case
10. Basal Cell Carcinomas Appearing During Immunotherapy for Reported Metastatic Basal Cell Carcinoma
11. Cutaneous Metastatic Sarcomatoid Renal Cell Carcinoma and Hand-Foot Syndrome
15A. Stewart-Treves Syndrome at site of Cavernous Lymphangioma
15B. Unusual Pigmented Angiosarcoma With Epithelioid Features Simulating Melanoma
16. Glomangiosarcoma
17. Cutaneous Endometriosis of the Umbilicus (Villar’s Nodule)
18. Tattoo reaction with pseudo-epitheliomatous hyperplasia to red pigment
20. Migrating Filler
21B. Bullous Morphea
24. Psoriasiform Dermatitis Secondary to Pembrolizumab Therapy
29A. Atypical (persistent) Eruption of Adult-onset Still’s Disease
29B. Adult Onset Still’s Disease with Macrophage Activation Syndrome
30. Symmetrical Drug Related Intertriginous And Flexural Exanthema (SDRIFE)
32. Nocardiosis
33. Dysmorphic Trichophyton rubrum Infection Simulating Blastomycosis
Case 1: Peripheral T-cell lymphoproliferative disorder, NOS CD4-/CD8-/BF-1+
Caregivers: Patrick Henry McDonough, MD, Etan Marks, DO, Clay J. Cockerell MD; Dallas, TX.
History: A 56 year old woman presented with a 4 month history of ulcerated plaques and nodules of the trunk and extremities. She complained of no “B” symptoms. She had a past medical history of fibromyalgia, depression, asthma, and hypertension. She is currently on Buspirone, Gabapentin, Losartan, Pantoprazole, and Venlafaxine.
Physical Examination: Scattered red papules and plaques with focal areas of necrosis and ulceration on the back of the neck, mid-back and legs.
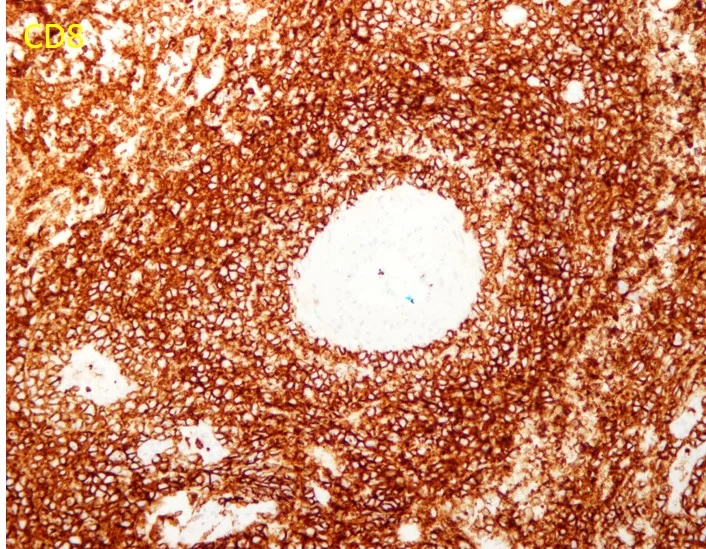
Histopathology: Band-like papillary dermal lymphoid infiltrate with epidermotropism comprised of large lymphocytes with irregular nuclei. The cells stained positively for CD3 and Beta-F1, but were mostly negative for CD4, CD8, CD30, and CD56. T-cell receptor gene rearrangements were positive for a clonal gamma T cell gene rearrangement.
Laboratory Data: Whole blood flow cytometry showed a CD4/CD8 ratio of 4.5. A Leukemia/Lymphoma Panel showed no abnormality. Serum T-cell receptor gamma gene rearrangement was negative. A CBC showed a WBC of 11.8 (slightly elevated) and platelet count of 456. A CMP showed no abnormalities. HIV and RPR tests were negative. However, PET/CT showed innumerable FDG-avid cutaneous lesions although no FDG-avid lymph nodes.
Clinical Course: The patient was treated with Prednisone 60mg daily and then tapered off as there was no improvement after 3 weeks. Methotrexate was added at a dosage of 17.5mg weekly. The patient continues to develop new lesions with persistence of older lesions.
Diagnosis: CD4-/CD8- T-cell Lymphoproliferative Disorder (Probably Mycosis Fungoides or Primary Cutaneous CD8+ Aggressive Epidermotropic Cytotoxic T-cell Lymphoma)
Points of Emphasis: The histopathological features of skin with prominent epidermotropism of atypical lymphocytes with clinically persistent plaques leads to a differential diagnoses that includes mycosis fungoides, primary cutaneous γ/δ T-cell lymphoma and primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. Often, these lesions can be differentiated by immunophenotyping with MF mostly showing a CD3+/CD4+/CD8-/BF-1+ phenotype, primary cutaneous γ/δ T-cell lymphoma showing a CD3+/CD4-/CD8-/BF-1- phenotype, and primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma showing a CD3+/CD4-/CD8+/BF-1+ phenotype. However, all three of these entities can show CD3+/CD4-/CD8-.
CD4/CD8 double-negative type MF has been commonly reported and has an indolent clinical course like that of typical MF. Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma can be CD4-/CD8- in some cases. Finally, γ/δ T-cell lymphoma is usually CD4-/CD8-, but shows negativity for BF-1.
Our case showed a CD4-/CD8-/BF-1+ immunophenotype which excludes the diagnosis of a γ/δ T-cell lymphoma. However, the clinical course appears to be more aggressive due to the persistent plaques and multiple cutaneous FDG avid lesions. Therefore, an unusual CD8- primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma seems to be more likely than a CD4-/CD8- mycosis fungoides.
References:
1. Hodak E, David M, Maron L, Aviram A, Kaganovsky E, Feinmesser M. CD4/CD8 double-negative epidermotropic cutaneous T-cell lymphoma: an immunohistochemical variant of mycosis fungoides. J Am Acad Dermatol 2006; 55: 276–284. 2. Toshinari Miyauchi, Riichiro Abe, Yusuke Morita, et al. CD4/CD8 Double-negative T-cell Lymphoma: A Variant of Primary Cutaneous CD8+ Aggressive Epidermotropic Cytotoxic T-cell Lymphoma? Acta Dermato-Venereologica. 2015;95(8):1024-1025.










Case 2: Blastic Plasmacytoid Dendritic Cell Neoplasm- An Underrecognoized but Deadly tumor
Caregivers: Aditya Sood UG-2, Dipti Anand MD; Smyrna, GA; Atlanta, GA
History: A 72 year old Caucasian male presented with acute onset of non-palpable, purpuric patches on left anterior proximal upper arm. This was clinically thought to be a bruise by physicians in an emergency room. The eruption did not resolve for ~2 months, and the patient was referred to dermatology.
Physical Examination: Physical examination showed an ecchymotic, bruised appearance of left arm with pigmented, purpuric patch and plaque. Clinical differential diagnoses included ecchymosis versus malignant melanoma.
Histopathology: A punch biopsy showed diffuse effacement of the dermal architecture with a dense perivascular, periadnexal and interstitial infiltrate of atypical lymphoid cells with hyperchromatic nuclei and inconspicuous nucleoli extending into the subcutis. Associated areas of dermal hemorrhage were seen. Immunophenotypically, the infiltrate was composed of CD45+ CD3- CD5- CD20- CD4+ CD8- CD56+ CD123+ TCL-1+ lymphoid cells, consistent with Blastic Plasmacytoid Dendritic cell neoplasm (BPDCN).
Clinical Course: Patient was referred to a tertiary medical care center for work-up for systemic involvement and chemotherapy.
Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN).
Points of Emphasis: Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) is a very clinically aggressive tumor derived from the precursors of plasmacytoid dendritic cells. It accounts for < 1% of all acute leukemia cases & 0.7% of cutaneous lymphomas. The male to female ratio for this tumor is ≥ 3:1. Most patients are elderly (mean age of 67 years at presentation).
Almost all cases have cutaneous lesions at presentation. Lesions can be solitary or multiple, presenting as, erythematous, violaceous or red-brown-nodules, tumors or plaques, with often a bruise-like appearance. There is a high frequency of bone marrow, peripheral blood (60–90%) and nodal (40–50%) involvement with leukemic dissemination.
In most cases, the neoplasm is confined to the skin at presentation. However, leukemic spread after variable, usually short, period of time is the rule, indicating that the primary cutaneous cases most likely represent “aleukemic” phase of leukemia cutis.
Initial responses to treatment are relatively common, but most patients relapse within a short space of time. It has a poor prognosis with median survival time of only 12–14 months (estimated 5-year survival is 0%). In 10–20% of cases, BPDCN is associated with, or develops into, a myelodysplastic syndrome, myelomonocytic leukemia or acute myeloid leukemia.
It is important to remember this very aggressive lymphoma with a high frequency of leukemic dissemination. Skin lesions are most often the first presenting manifestation of the disease. Cutaneous lesions can be bruise-like or pigmented simulating melanoma. Histology can pose a diagnostic challenge. Because of its aggressive behavior, it is important to correctly diagnose it in a timely fashion for proper clinical management.
References: 1. Wasif, R., Zhang, L., Horna, P., Sokol, L., Blastic Plasmacytoid Dendridic Cell Neoplasm: Update on Molecular Biology, Diagnosis, and Therapy, Cancer Control 291, Vol 21 No 4. , Oct. 2015 . 2. Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC Press; 2008.






Case 3A: A Unique Case of Extranodal NK/T-Cell Lymphoma Mimicking Mycosis Fungoides
Casregivers: Michael James Davis, BMus, Dipti Anand, MD; Atlanta, GA
History: An 80-year-old Caucasian gentleman with no significant past medical or drug history presented with multiple acute and rapidly progressing lesions. No systemic symptoms such as fever, malaise or weight loss were reported.
Physical Exam: Exam revealed several 1-3 cm well demarcated, erythematous and violaceous plaques on both sun exposed and non-sun exposed aspects of the patient’s extremities and abdomen. No facial involvement was noted. Deep punch biopsies of the right anterior thigh, right posterior arm, and right anterior arm were performed.
Laboratory Data: ANA -, Anti Ro -, Anti La -, ESR 2.
Histopathology: The sections showed a dense superficial and deep perivascular, perifollicular, and interstitial lymphoid infiltrate extending into the subcutaneous tissue. The lymphocytes were pleomorphic, intermediate to large, with irregular chromatin distribution and scattered mitoses. The overlying epidermis demonstrated subtle interface vacuolization with scattered single and collections of lymphocytes, suggestive of epidermotropism. There was no significant necrosis or vasculitis, but extravasation of erythrocytes with riming of fat by atypical lymphocytes was seen.
The histologic differential diagnoses included plaque and tumor stage mycosis fungoides, pseudolymphomatous lupus panniculitis, cytotoxic subcutaneous panniculitis-like-T-cell lymphoma and primary cutaneous gamma/delta T cell lymphoma.
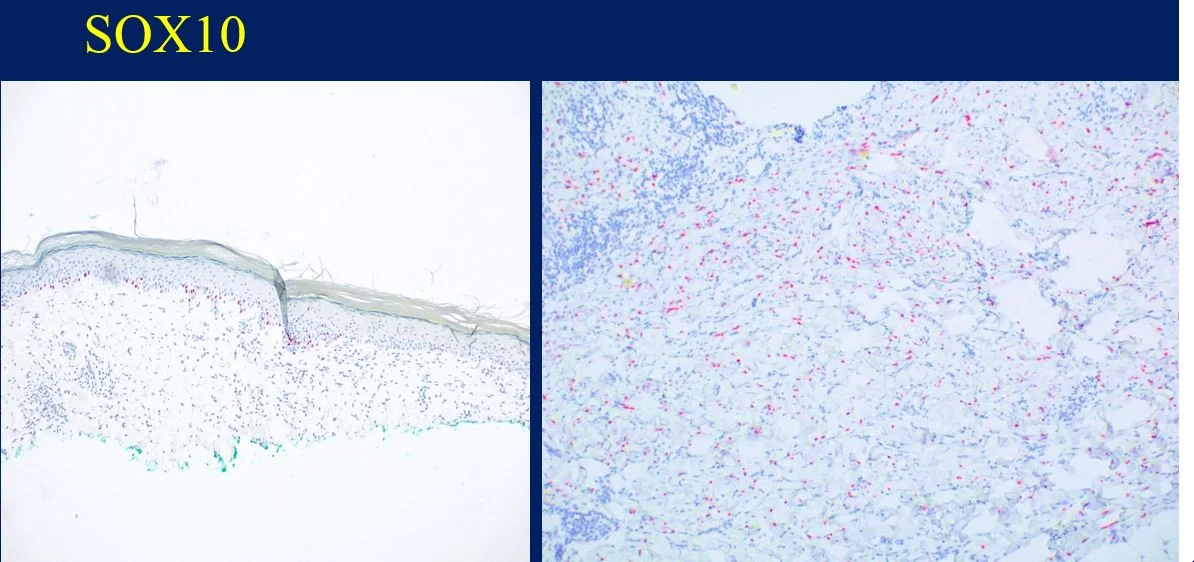
Immunohisochemical evaluation revealed tumor cells to be positive for CD2, CD3, CD56, TIA-1, and granzyme B, and negative for CD5, CD7, CD4, CD8, CD20, and CD123. Additionally, CD 30, Beta FI Gamma M1 were also negative. EBV-encoded RNA (EBER)-1 in situ hybridization was positive for EBV mRNA. RT-PCR T-Cell gene arrangement study was positive.
The histology and staining profile were consistent with EBV positive Extranodal NK/T-Cell Lymphoma.
Clinical Course: Systemic workup for nasopharyngeal and extra nodal site involvement was recommended at a tertiary care center.
Diagnosis: Extranodal NK/T-Cell Lymphoma.
Points of Emphasis: Beyond the classic presentation of an ulcerated mid facial tumor, previously coined “lethal midline granuloma,” Extranodal NK/T-Cell Lymphoma requires a high index of suspicion for accurate diagnosis. Following the nose and upper aerodigestive tract, the skin is the most commonly involved site. Lesions are typically plaques or nodules, at times ulcerated; vasculitis and panniculitis presentations can also occur. The infiltrate is usually angiocentric and may be angiodestructive with variably sized tumor cells.
Immunophenotypically, the tumor cells express CD2, CD3ɛ, CD56, TIA-1, and granzyme B. EBER in situ hybridization is the most reliable test for confirming the presence of Ebstein-Barr Virus which is detected in almost all cases. Extranodal NK/T-cell lymphoma presenting in the skin is a highly aggressive tumor with a median survival of less than 15 months.
Our patient presented with a unique clinical and histologic manifestation of this uncommon lymphoma. Clinically, the lesions mimicked lupus panniculitis and mycosis fungoides, and lacked the tumor nodules and ulceration, often seen in this lymphoma. The histology also posed a challenge, simulating mycosis fungoides and pseudolymphomatous lupus erythematosus. The lymphoma lacked the necrosis, prominent angiocentricity and angiodestruction, and high mitotic rate with apoptosis, which is classic for this neoplasm. Epidermotropism as noted in our case is seen in ~30% of these tumors.
It is important for both clinicians and pathologists to be aware of this uncommon cutaneous T-cell lymphoma, which can mimic mycosis fungoides both clinically and histologically. Awareness of this entity is important for its prompt recognition and treatment.
References: 1. Park, S, Ko, YH. “Epstein-Barr virus-associated T/natural killer-cell lymphoproliferative disorders.” J Dermatol. 2014;41:29-39. 2. Peck, T, Wick, M. “Primary cutaneous natural killer/T-cell lymphoma of the nasal type: a report of 4 cases in North American patients.” Ann Diagn Pathol. 2015;19:211-215. 3. Takata, K et al. “Primary Cutaneous NK/T-cell Lymphoma, Nasal Type and CD56-positive Peripheral T-cell Lymphoma: A Cellular and Clinicopathologic Study of 60 Patients from Asia.” Am J Surg Pathol. 2015;39:1-12.





















Case 3B: NK/T-Cell Lymphoma
Caregivers: Neeta Malviya BS, Juliet F. Gibson, MD, Yevgeniya Byekova Rainwater, MD; Dallas, TX
History: A 28-year-old male with a medical history of HIV infection presented with a 1-month history of a steadily enlarging, firm painful lesion on the right posterior shoulder. The lesion was initially diagnosed as an abscess with surrounding cellulitis and treated with multiple I&Ds in the emergency department with no improvement and progressive swelling of the right upper extremity despite numerous systemic antibiotics.
Physical Examination: Physical examination revealed a 15 cm yellow-brown, verrucous plaque on the right posterior shoulder with surrounding violaceous erythema and induration. Within 1 week, plaque ulcerated and developed an eschar.
Laboratory Data: Baseline laboratory studies revealed white blood cell count of 13 (reference range 4.22-10.33 cells/microliter), hemoglobin 11.5 (reference range 13.2-16.9g/dL), CD4 count of 507 (reference range 212-1403 cells/mm3), HIV viral load 111 copies/mL, negative blood and urine cultures, and wound cultures growing rare Acinetobacter lwoffi.
Histopathology: Histopathologic examination revealed an angiocentric and dermal proliferation of markedly atypical lymphoid cells with numerous mitoses and apoptotic bodies along with broad zones of necrosis. Atypical lymphoid cells highlighted with CD2, CD3, CD8, CD30, and CD56. CD20, PAX5, and CD4 stains were negative. In situ hybridization for Epstein Barr virus revealed positivity within nearly all atypical lymphoid cells. An ultrasound-guided lymph node biopsy of the right axilla revealed morphologic and immunophenotypic findings consistent with a NK/T cell lymphoma.
Clinical Course: Computed tomography of the chest revealed a multicentric lymphoma producing a large right axillary mass and enlarging left paraspinous musculature. Nasal computed tomography did not reveal any masses. Bone marrow biopsy was found to be benign. Computed tomopgraphy of the abdomen and pelvis revealed an infiltration of the right gluteus medius and minimus muscles as well as the left paraspinous muscles at the level of the thoracolumbar junction. Overall lymphoma involvement included lymph nodes, multiple soft tissue sites including the right shoulder, back, left thigh and left knee. Two cycles of SMILE (steroid (dexamethasone), methotrexate, ifosfamide, L-asparaginase, and etoposide) chemotherapy were administered as per treatment protocol and the patient tolerated these well. This was followed with EPOCH therapy (Rituximab, etoposide, prednisolone, oncovin, cyclophosphamide, hydrocydanorubicin), which the patient also tolerated well and resulted in improvement of the shoulder lesion.
Diagnosis: NK/T-cell lymphoma.
Points of Emphasis: The cutaneous lesions from NK/T-cell lymphoma can often be initially mistaken for cellulitis as in the case of our patient. In a study of 41 cases of cutaneous extra-nodal NK/T-cell lymphoma, skin lesions presented in 3 different categories, nodular lesions, cellulitis or abscess-like swellings, and erythematous to purpuric patches (1). In the histopathologic analysis of these categories, it was found that tumor cell infiltration density was greater in cellulitis-like lesions or nodular lesions as compared to erythematous to purpuric patches. Both cellulitis-like lesions and nodular lesions are also more likely to display angiocentricity as was the case in our presented patient (1). The original cutaneous presentation in our reported case was of a cellulitis appearing lesion on the right arm, this later became necrotic in appearance and formed an eschar. The formation of an eschar is noted in one other case report of a 14-year-old boy with cutaneous involvement in NK/T-cell lymphoma (2). In distinguishing this lesion from cellulitis, failure of antibiotic treatment, progressive and widespread involvement of the cellulitis along with profound ulceration should prompt the clinician to explore an alternative diagnosis as well as suggest the possibility of NK-T cell lymphoma (3).
References: 1. Lee WJ, Lee MH, Won CH, Chang SE, Choi JH, Moon KC, et al. Comparative histopathologic analysis of cutaneous extranodal natural killer/T‐cell lymphomas according to their clinical morphology. Journal of cutaneous pathology. 2015. 2. Xiao HJ, Li J, Song HM, Li ZH, Dong M, Zhou XG. Epstein-Barr Virus-Positive T/NK-Cell Lymphoproliferative Disorders Manifested as Gastrointestinal Perforations and Skin Lesions: A Case Report. Medicine. 2016;95(5):e2676. 3. Kim SH, Seon HJ, Choi YD, Yun SJ. Clinico-radiologic findings in primary cutaneous extranodal natural killer/t-cell lymphoma, nasal type mimicking cellulitis of the left arm. Iranian journal of radiology : a quarterly journal published by the Iranian Radiological Society. 2015;12(1):e12597.











Case 4A: Congenital Leukemia Cutis as Presenting Feature of Acute Myeloid Leukemia
Caregivers: Caroline Lee, MD, Matthew LaCour, MS4, Jill Fruge, MD, Beverly Ogden, MD; New Orleans, LA, Baton Rouge, LA, Galveston, TX
History: ER is a 7 day old female infant who presented with non-pruritic blue to purple lesions over her trunk, neck, and extremities, first noticed at birth. She was born at 40 weeks and 3 days via Caesarean section after failed induction. This was her mother’s first pregnancy and pregnancy was without illnesses or complications. The patient was feeding well and having normal urine and stool output.
Physical Examination: Vitals: Temp: 98.5 degrees F, BP: 95/59mmhg, Pulse: 164, RR: 54, SpO2: 96%; Lymph nodes: negative for cervical, axillary, or inguinal lymphadenopathy
Abdomen: negative for hepatomegaly and splenomegaly
Skin: 2-4 mm red-violaceous papules distributed over abdomen, back, extremities and diaper area; violaceous nodules to right neck and on right foot
Laboratory Data: WBC count: 15.9 1000/ul (WNL), HGB: 13.7 gm/dl (WNL), HCT: 41.5 %, MCV: 102 fl (WNL), PLT count: 473 1000/ul (elevated), Differential: 50% neutrophils, 32% lymphocytes, 15% monocytes (elevated), 3% eosinophils, 1% basophils. No circulating malignant cells observed. CMP was unrevealing. LDH: 595 IU/L (elevated). Blood cultures were negative. Serologic studies for toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus were negative.
Histopathology: H&E: Malignant infiltrate of immature cells. Several immunostains were performed including CD45, CD34, CD117, myeloperoxidase, S100, CD68, CD79A, CD3, desmin, cytokeratin A, CD31, and CD43. The malignant cells are positive for CD34, CD68, CD43 and are negative for the other markers tested. These findings favor leukemia cutis/myeloid sarcoma over other small blue cell tumors.
Clinical Course: The hematology-oncology service was initially consulted and peripheral blood smear was found to be unremarkable. An infectious disease work-up was also unremarkable. The dermatology service was then consulted and the biopsy specimen was concerning for myeloid leukemia cutis. The patient was referred to St. Jude Children’s Hospital where further work-up was performed including a bone marrow aspirate which was consistent with acute myeloid leukemia. Cytogenetic analysis showed a CIC mutation in 15/20 metaphases. She completed Induction I chemotherapy including cytarabine, daunorubicin, etoposide (ADE) and Induction II chemotherapy with cytarabine, daunomycin, etoposide and intrathecal methotrexate, hydrocortisone, cytarabine. She is currently in remission and is completing the consolidation phase of chemotherapy
Diagnosis: Congenital leukemia cutis as the presenting feature of Acute Myeloid Leukemia
Points of Emphasis: Infants born with a cutaneous eruption characterized by deep red to blue macules, papules and nodules are characterized as having a “blueberry muffin syndrome.”1-3 The most common causes of a blueberry muffin eruption include dermal erythropoiesis and neoplastic infiltrative diseases. An infant presenting with this eruption should undergo an extensive work-up to determine the underlying cause.2, 3 Hematopoiesis typically takes place in the skin from the first through fifth months of gestation.1, 2 Occasionally dermal erythropoiesis takes place during the neonatal period which may be due to an intrauterine viral infection, most commonly rubella, toxoplasmosis, or parvovirus, or due to congenital hematologic dyscrasias, including hereditary spherocytosis, twin-twin transfusion syndrome, or hemolytic disease of the newborn. Infants should also be evaluated for neoplastic infiltrate diseases including metastatic neuroblastoma, histiocytic disorders and congenital leukemia.1-4
Congenital leukemia encompasses all cases diagnosed from birth to 6 weeks of life with acute myeloid leukemia (AML) denoted as the most common type. The etiology of congenital leukemia is unknown, but it has been associated with maternal exposure to radiation, bioflavonoid, tobacco and illicit drugs, topoisomerase II inhibitors, such as coffee, tea, cocoa, wine (catechins), and soy products (genistein), high birth weight (>4000 g), high levels of insulin-like growth factors, and disorders such as Down syndrome. Infants with congenital leukemia may present with varying signs and symptoms, with leukemia cutis presenting in 25-30% of cases.3, 4
Leukemia cutis (LC) represents a cutaneous neoplastic leukocytic infiltrate and may be the presenting sign of an underlying leukemia.1,3,5 Patients with LC at birth can have variable cutaneous manifestations but most commonly present with multiple firm blue, red, or purple papules, nodules, or plaques that give rise to a “blueberry muffin” appearance.1, 3 Leukemic skin symptoms are most commonly noted in patients with AML that have a prominent monocytic or myelomonocytic component. Rarely, cutaneous involvement by a leukemic infiltrate can occur without concurrent bone marrow or peripheral blood involvement by acute leukemia, which is referred to as aleukemic LC.6
Diagnosis of leukemia cutis is confirmed by the pattern of skin infiltration, specific histomorphology, and the immunohistochemical staining pattern of the cells. Histology should be correlated with the clinical presentation and peripheral blood and bone marrow findings.7 LC may show different patterns of infiltration. The most common presentation includes a dense diffuse dermal infiltrate of pleomorphic leukemic cells arranged in a linear array between collagen bundles in the reticular dermis. The infiltrate may also commonly present in a periadnexal or perivascular fashion. Typically, infiltrates diffusely extend into the dermis and subcutis but spare the epidermis resulting in a Grenz zone. Malignant myeloid cells are numerous, and nuclear characteristics may range from markedly atypical folded or reniform nuclei to monomorphous and cytologically bland nuclei.1, 3, 4 Immunohistochemical analysis is required for confirmation. Malignant cells are negative for CD3 and CD20, effectively ruling out a lymphocytic process. Most commonly in myeloid leukemia cutis, cells will be CD43+/myeloperoxidase (MPO)+ or CD43+, MPO-, CD68+, CD56-, CD117-.1,5 A recent cohort study by Cronin et al detailing the approach to diagnosis of myeloid leukemia cutis found that CD43 was positive in 97%, myeloperoxidase was positive in 42%, CD68 was positive in 94%, CD163 was positive in 25%, and CD56 was positive in 47% of those diagnosed with myeloid LC.5 They point out that when myeloperoxidase staining was negative CD43 and CD68 were both positive.5
Since LC indicates a systemic leukemia, treatment of the underlying leukemia typically resolves the cutaneous eruptions of LC. Referral and close follow up with Hematology-Oncology should be implemented. Unlike LC in adults that denotes extramedullary involvement and poor prognosis, LC secondary to congenital leukemia does not denote a worse prognosis. Regardless, congenital leukemia due to AML has a poor prognosis with some reports of survival rates as low as 26%, thus rapid diagnosis and treatment is essential to improve mortality.3,4
References: 1. Weedon J. Weedon’s Skin Pathology. 4th edition. Charlottesville (VA): Elsevier Saunders; 2016. 2. Gottesfeld E, Siverman R, Coccia P, Jacobs G, Zaim M. Transient blueberry muffin appearance of a newborn with congenital monoblastic leukemia. J AM Acad Dermatol. 1989;21:347-51. 3. Zhang I, Zane L, Braun B, Maize J, Zoger S, Loh M. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006 Feb; 54(2 Suppl):S22-7. 4. Choi JH, Lee HB, Park CW, Lee CH. A Case of Congenital Leukemia Cutis. Annals of Dermatology. 2009;21(1):66-70. doi:10.5021/ad.2009.21.1.66.





Case 4b: Leukemic Infiltrate Secondary to Chronic Myelogenous Leukemia
Caregivers: Carole Bitar, MD, Carole Bitar, MD; Jacqueline Witt, MS; Jalal Maghfour, MS; New Orleans, LA
History: This is a 45-year old woman with past medical history of chronic myelogenous leukemia (CML) on nilotinib that was admitted to the hospital for progressive swelling and severe pain of her left leg. The process started 6 days previously when she initially noticed a “knot” under her skin that was itchy then it became red and swollen. She experienced pain and burning sensation in that area. The patient noted that she experienced chills and subjective fever at home. She denied any trauma. Doppler ultrasound was unremarkable and there were no signs of DVT. She was started on clindamycin for presumed cellulitis and started to improve with decrease redness and swelling although there was a persistent subcutaneous nodule on the left leg. The subcutaneous nodule had a greenish discoloration. The patient reported that additional green subcutaneous nodules appeared on the right arm. She had a past history of leukemia cutis with abundant blasts in 2017 for which she was treated with induction chemotherapy. On this admission, she was started on hydroxyurea and nilotinib.
Current Medications: Clindamycin, hydroxyurea, nilotinib, pantoprazole.
Physical Examination: There is an erythematous plaque over the left shin, tender to touch and warm with a centrally located greenish subcutaneous nodule. Two subcutaneous greenish nodules over the right arm were present.
Laboratory Data: Two punch biopsies were performed over the left leg for hematoxylin and eosin stain and for flow cytometry. Laboratory work up was significant for very high WBC of 167x103. Bone marrow biopsy showed chronic myeloid leukemia with no blasts.
Histopathology: A punch biopsy showed an atypical perivascular infiltrate comprised of neutrophilic, eosinophilic, and mononuclear cells compatible with immature myelocytes involving the dermis and subcutis with hemorrhagic lobular panniculitis. The infiltrate was not blastic but was compatible with leukemic infiltrate secondary to chronic myelogenous leukemia. The majority of perivascular cells stained positive for CD68. Myeloperoxidase stained numerous cells compatible with myelocytes. ERG immunostain confirms the presence of endothelial cells.
Flow cytometry: Nucleated cellularity consists predominantly of granulocytic cells. While there is a minute population (1.6%) of CD34+ cells falling within a typical blast gate by CD45/side scatter, these do not form a discrete cluster on examined histograms. It is noted that this patient's prior cutaneous leukemia involvement reportedly manifested as a mixture of mature and immature cells; therefore, morphologic correlation is essential.
Clinical Course: The patient continues to be followed by hematology/oncology and maintain current medication regimen. She is hesitant to undergo the recommended induction chemotherapy.
Diagnosis: Leukemic infiltrate secondary to chronic myelogenous leukemia.
Points of Emphasis: Leukemia cutis is a broad diagnosis for all cutaneous manifestations of leukemia caused by either myeloid or lymphocytic infiltrates to the skin. Although most frequently associated with acute myeloid leukemia (AML), it has also been described in chronic myeloproliferative disorders. Typically, a diagnosis of leukemia predates skin involvement, but rarely, skin disease may be the first indication of systemic disease. Roughly 10-15% of patients with acute myeloid leukemia are diagnosed with leukemia cutis. In comparison, up to 25-30% of infants with congenital leukemias have skin involvement, the majority of which are associated with AML. Almost half of adult patients with acute myelomonocytic and monocytic subtypes of AML will develop skin manifestations1. Between 4 and 20% of chronic lymphocytic leukemia/small lymphocytic lymphoma patients have cutaneous involvement and 20-70% of mature T-cell leukemias involve the skin. Rarely do precursor B- or T-cell lymphoblastic leukemias and lymphomas or plasma cell myelomas involve the skin1.
Patients present with violaceous and erythematous papules, nodules, and plaques. Often manifesting at points of previous or current inflammation, the lower extremities are most frequently affected1. Rarely, clinical features described as leonine faces have been reported in a subset of leukemia cutis patients as well as panniculitis mimicking erythema nodosum. Close to a third of children with leukemia cutis will demonstrate the classic “blueberry muffin” appearance. Higher levels of serum lactate dehydrogenase and ß2-microglobulin have been reported in patients with leukemia cutis versus those with leukemia without cutaneous manifestations. No other clinical or demographic associations have been reported1.
Histopathologically, leukemia cutis demonstrates perivascular and periadnexal infiltrates of the dermis and subcutaneous tissue without involvement of the upper papillary dermis. Immunophenotyping is crucial to diagnosing leukemia cutis, as pathology is often non-specific. Important stains such as myeloperoxidase and lysozyme are also important for identifying myeloid cells2. The neural cell adhesion molecule CD56 is important for identifying AML, although its sensitivity has been debated. Other molecules such as CD68/KP-1 and CD117 are also valuable in identifying AML.
Myeloid sarcoma, also known as granulocytic sarcoma, is a cutaneous manifestation of malignant myeloid cells and therefore a subset of leukemia cutis. It is also classically referred to as “chloroma” because of the greenish hue from the enzymatic function of myeloperoxidase3. Myeloid sarcoma is most commonly associated with AML and occurs in 2-3% of those patients3. It very rarely appears in CML patients mainly in the setting of blastic transformation.
The prognosis and treatment of leukemia cutis relies on addressing the underlying disease. Many reports suggest that by the time the disease manifests itself in the skin, the patient’s prognosis is poor. It has been reported that 88% of patients will die within 1 year of diagnosis of leukemia cutis1. This poor prognosis has been attributed to an associated blast transformation and crisis indicative of disease progression and often accompanying the presentation of leukemia cutis. Interestingly, patients with leukemia cutis secondary to a congenital leukemia do not have such a poor prognosis.
References: 1. Cho-Vega JH, Medeiros LJ, Prieto VG, Vega F. Leukemia cutis. Am J Clin Pathol. 2008 Jan;129(1):130-42. 2. Wang YN, Lian CG, Hu N, Jin HZ, Liu YH. Clinical and pathological features of myeloid leukemia cutis. An Bras Dermatol. 2018;93(2):216-21. 3. Vasconcelos ERA, Bauk AR, Rochael MC. Cutaneous myeloid sarcoma associated with chronic myeloid leukemia. An Bras Dermatol. 2017;92(5 Suppl 1): 50-2.









Case 5A: Atypical Spitz Nevus in a 3-Year-Old
Caregivers: Margaret Snyder, Weston Wall MD, Daniel J. Sheehan MD, Loretta S. Davis MD; Augusta, GA
History: A 3-year-old girl with no family history of skin disease presented to dermatology with a 2-year history of a growth on her left earlobe. The mother stated that the growth periodically changed in size, sometimes enlarging and other times shrinking.
Physical Examination: Physical examination revealed a flesh-colored 3x4mm papule on the left earlobe.
Histopathology: Initial Shave Biopsy: Compound proliferation of enlarged spitzoid melanocytes, involving the deep edge of the biopsy. Molecular analysis of the specimen via the Affymetrix Oncoscan Microarray system revealed mosaic loss of chromosomes 9 and 14, a finding inconsistent with pediatric spitzoid melanoma and supportive of classification as an “atypical spitz tumor.” Re-excision was recommended. Final resected specimen: Pan-Melanoma cocktail revealed microscopic foci of residual compound Spitz tumor, with negative resection margins.
Clinical Course: Plastic surgery excised the residual lesion, achieving negative margins. Patient has since been seen by dermatology and plastic surgery, with well-healing cicatrix and negative recurrence at 11 months.
Diagnosis: Atypical Spitz Nevus
Points of Emphasis: Spitz nevi are uncommon melanocytic neoplasms seen in the pediatric population. These lesions are classified on a spectrum of increasing severity from typical Spitz nevus to atypical Spitz nevus to atypical Spitz tumor to spitzoid melanoma. Due to significant dermoscopic and histopathological overlap throughout the spectrum, precise diagnosis remains difficult and unstandardized. Intermediate categories of “atypical Spitz nevi” and “atypical Spitz tumor” show more marked atypical features than typical Spitz nevi, but do not meet the criteria to be classified as spitzoid melanoma. The malignant potential of intermediate categories is therefore uncertain, resulting in a lack of consensus among dermatologists, plastic surgeons, and pathologists regarding treatment. Indeed, there currently exists no algorithmic approach to atypical Spitz nevi or atypical Spitz tumor.
References: 1. Abboud J, Stein M, Ramien M, Malic C. The diagnosis and management of the Spitz nevus in the pediatric population: a systematic review and meta-analysis protocol. Syst Rev. 2017 Apr 13;6(1):81 2. Ferrara G, Gianotti R, et al. Spitz nevus, Spitz tumor, and spitzoid melanoma: a comprehensive clinicopathologic overview. Dermatol Clin. 2013 Oct;31(4):589-98, viii. doi: 10.1016/j.det.2013.06.012. 3. Metzger A, Kane A, et al. Differences in Treatment of Spitz Nevi and Atypical Spitz Tumors in Pediatric Patients Among Dermatologists and Plastic Surgeons. JAMA Dermatol. 2013 Nov;149(11):1348-50. doi: 10.1001/jamadermatol.2013.4947.






Case 5B: Pigmented Spindle Cell (Reed’s) Nevus with Chromosomal Mutations
Caregivers: Osama Hashmi MPH, Jack Yu, MD, Daniel Sheehan, MD; Augusta, GA
History: A 13 year old Hispanic male presented to his pediatrician for an annual checkup. He has no significant medical, birth, or social history. He noted a raised black lesion on his right arm which has been enlarging over the past five years. There was no personal or family history of melanoma. Patient also denied any pain, itching or bleeding with the lesion.
Physical Examination: Physical exam revealed a 7mm irregular darkly pigmented papule on the right arm with an irregular border. The papule was excised by plastic surgery.
Laboratory Data: Not applicable.
Histopathology: There is a well circumscribed proliferation of moderately enlarged spindled melanocytes, arranged mostly as nets along the dermoepidermal junction. There is coarsely papillated epidermal hyperplasia. The junctional melanocytes have scant, pigmented cytoplasm and small monomorphous oval nuclei. Smaller, more oval and less pigmented cells are present in the subjacent papillary dermis. There is pigmentation of the basal layer and in subjacent melanophages. DNA analysis was performed using the Affymetrix Oncoscan Microarray system. BRAF600VE mutation was not identified on the specimen. Mosaic losses were identified on chromosomes 6 (6q22.1q27) and 15 (15q11.1q21.2) including MYB.
Clinical Course: No recurrence of the lesion has been noted.
Diagnosis: Pigmented Spindle Cell (Reed’s) Nevus with NTRK3 or ROS1 gene fusion
Points of Emphasis: Pigmented spindle cell nevus of Reed is a variant of Spitz nevus and can be clinically and histologically challenging to distinguish from melanoma. BAP1, ALK, NTRK1, NTRK3, BRAF, ROS1, and HRA have been associated with Spitz nevus and literature has shown these genetic mutations to be specific to spitzoid neoplasms, distinguishing them from nonspitzoid melanoma, blue melanocytic lesions, and deep penetrating nevi. In this case, a genetic mutation was identified on chromosomes 6 and 15. Chromosome 15 is a known location for the NTRK3 gene and chromosome 6 is a known location for the ROS1 gene. This case illustrates possible genomic mutation that can be found in this variant of Spitz nevus.
References: 1. Urso, Carmelo. "Spitzoid Neoplasms: Suggestions from Genomic Aberrations." Dermatopathology 5.1 (2018): 26-29. 2. VandenBoom T, Quan V, Gerami P, et al. Genomic Fusions in Pigmented Spindle Cell Nevus of Reed. American Journal Of Surgical Pathology [serial online]. August 2018;42(8):1042. 3. Lee CY, Sholl LM, Zhang B, et al. Atypical Spitzoid Neoplasms in Childhood: A Molecular and Outcome Study. Am J Dermatopathol. 2017;39(3):181-186. 4. Amatu, Alessio, Andrea Sartore-Bianchi, and Salvatore Siena. "NTRK gene fusions as novel targets of cancer therapy across multiple tumour types." ESMO open 1.2 (2016): e000023.



Case 6: A Case of Desmoplastic Malignant Melanoma: A Diagnostic Pitfall
Caregivers: Michael James Davis, BMus, Dipti Anand, MD; Atlanta, GA
History: A 66-year-old Caucasian woman presented for her routine physical examination and was found to have a lesion on her left upper arm. Physical examination and review of systems were otherwise negative.
Physical Exam: A 0.5 x 1.0 cm poorly defined, asymmetric pink plaque with irregular borders and subtle dotted vessels was present on the left upper arm. A shave biopsy with the clinical impression of basal cell carcinoma vs squamous cell carcinoma was performed.
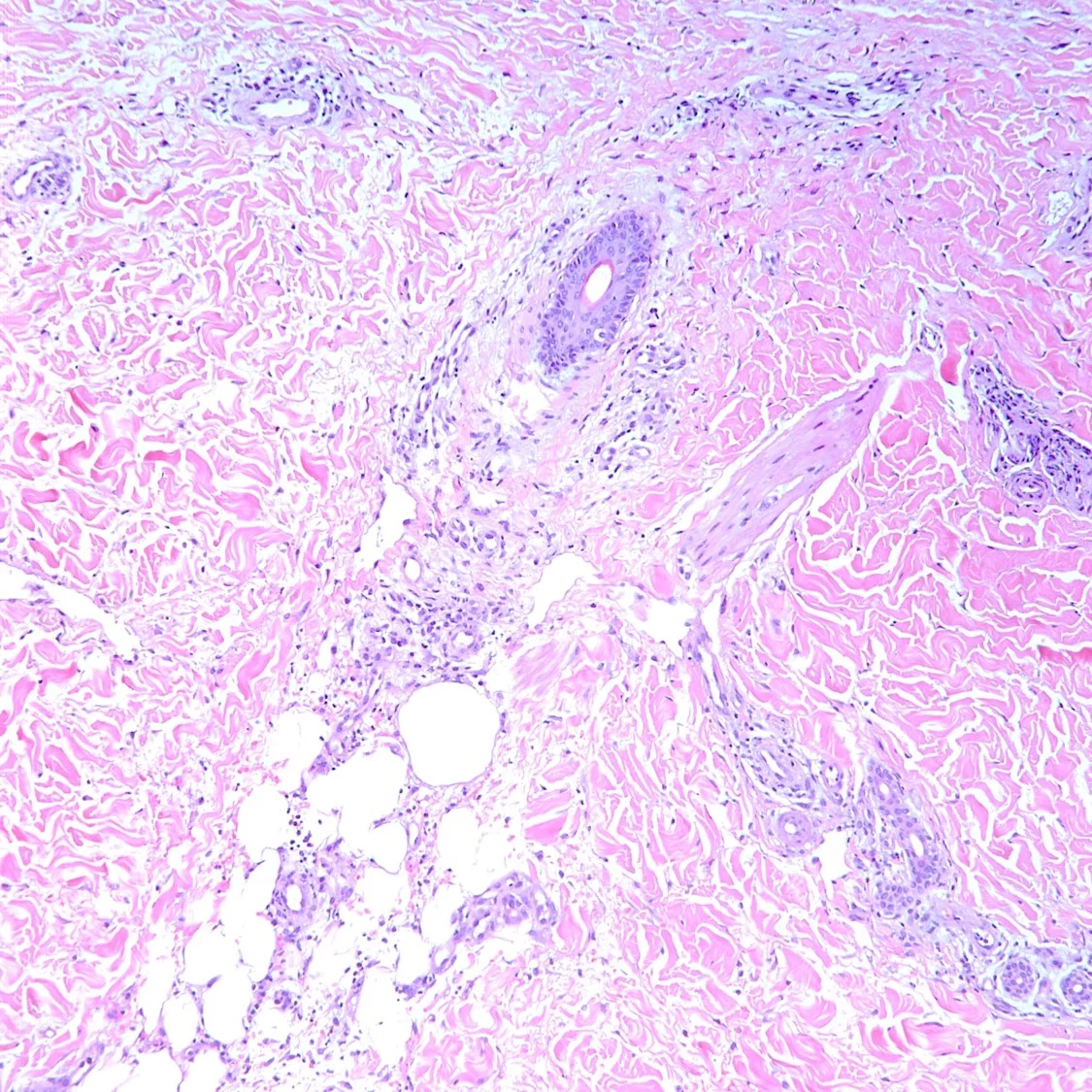
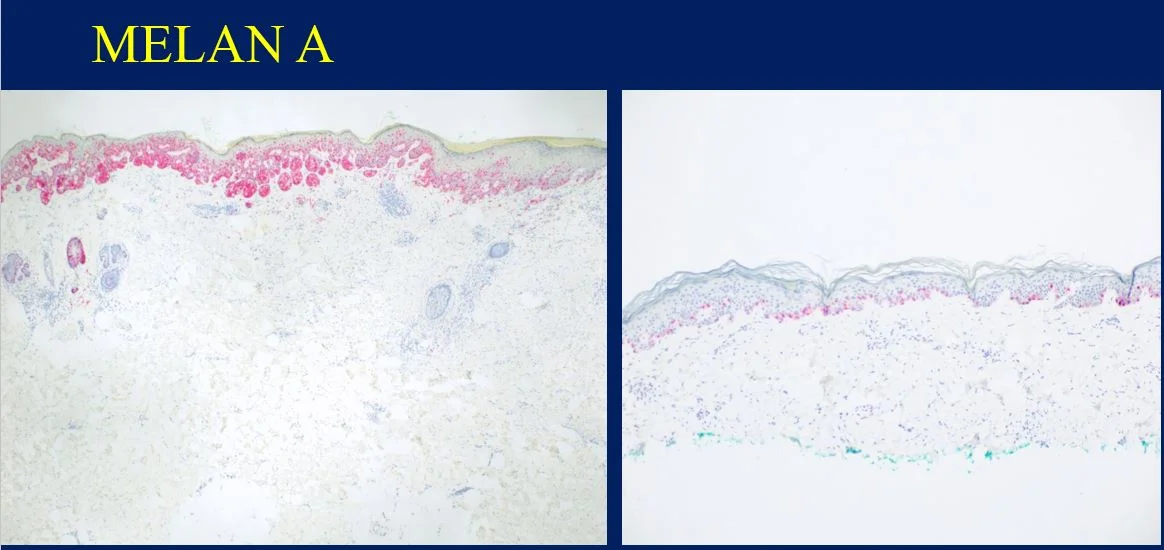
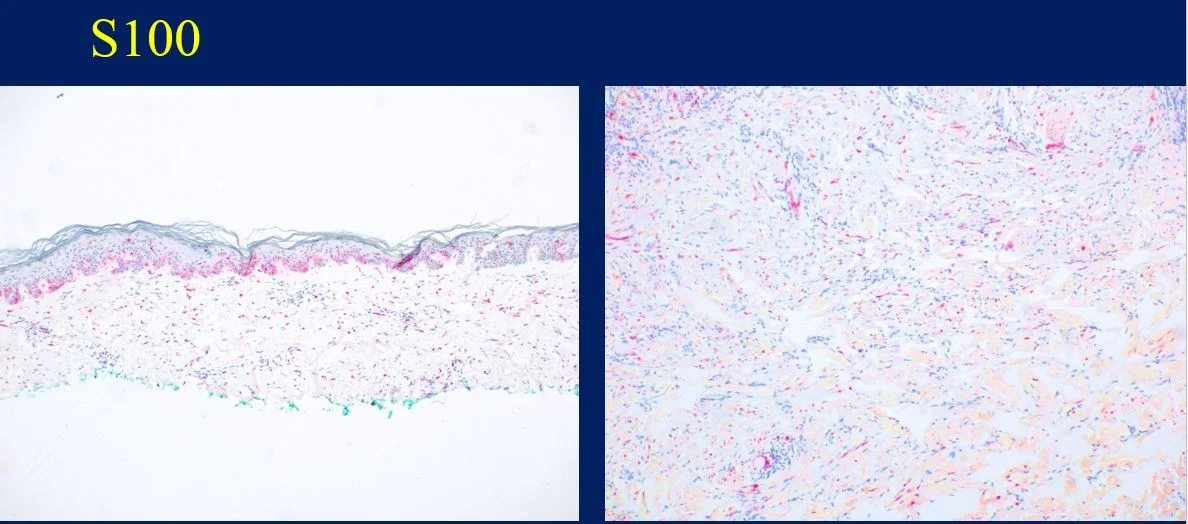
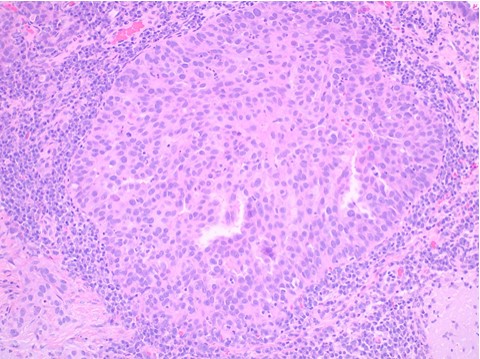
Histopathology: Histology showed dermal scarring with stromal perivascular lymphocytic inflammation, and slight junctional melanocytic proliferation. S-100, Melan-A and SOX-10 immunohistochemical markers highlighted the patchy atypical junctional melanocytic proliferation, with S-100 and SOX-10 also showing focal and weak staining of the dermal spindle cells. Additional information revealed no prior history of biopsy at this site. The pathology was signed out as “atypical compound melanocytic proliferation with scar” with recommendation for complete removal of the lesion and correlation with the residuum in the re-excision specimen.
A full thickness re-excision of the site showed, in association with scar of the previous procedure, classic appearing melanoma in-situ with lentiginous, contiguous junctional melanocytic proliferation of single and nests of enlarged, atypical melanocytes. In the dermis was a diffuse proliferation of cytologically banal, delicate spindle cells with mildly enlarged and slightly hyperchromatic nuclei and occasional dermal mitoses (~2/mm2). Associated trailing lymphoid aggregates were noted in the stroma. S-100 and SOX-10 diffusely highlighted the dermal melanocytic proliferation which was negative for Melan-A.
Diagnosis: Invasive Malignant Melanoma, Desmoplastic type, with a Breslow thickness of 2.1 mm, Clark’s level 4, and with a pathologic tumor stage of pT3a.
Points of Emphasis: Desmoplastic Melanoma (DM) is a rare cutaneous malignancy that proves a challenge for both clinicians and pathologists because of its subtle presentation and broad differential diagnosis. It is typically found on chronically sun-damaged skin of older individuals and has a male to female ratio of approximately 2:1. DM often presents as an amelanotic plaque or an ill-defined scar-like lesion that lacks an epidermal component; it can also be found in the background of other melanomas, most commonly lentigo maligna melanoma. Compared to conventional melanoma, DM has both a lower rate of nodal metastasis despite greater median tumor thickness at presentation and a relatively higher incidence of local recurrence.
Histologically, DM remains a challenging diagnosis and necessitates a high index of suspicion. Atypical spindle cells are found in a densely hyalinized collagenous stroma. There can be deep infiltration and invasion into the subcutis, and DM has a high propensity for perineural invasion. Trailing lymphoid aggregates can be a subtle clue in reaching the diagnosis. An important pitfall to appreciate is that DM can be negative for melanocytic markers including Melan-A, Mart-1, and HMB-45. In this case, the tumor was negative for Melan-A.
References: 1. Han, D et al. “Clinicopathologic Predictors of Survival in Patients with Desmoplastic Melanoma.” PLoS One. 2015;10:e0119716. 2. Chen LL, BA, Jaimes, N, Barker CA, Busam KJ, Marghoob, AA. “Desmoplastic melanoma: A review.” J Am Acad Dermatol. 2013;68:825-833.








Case 7: Ectopic Extramammary Paget’s Disease Presenting on the Face- An Extraordinary Case
Caregivers: Dipti Anand, MD, Aditya Sood, UG-3; Athens, GA and Atlanta, GA
History: An 81 year old Caucasian male with past medical history of gastric adenocarcinoma (diagnosed in March 2015), on systemic chemotherapy with capecitabine (Xeloda), presented with a pink plaque on his right cheek.
Physical Exam: A 2 cm erythematous, scaly plaque was noted on the right cheek. The clinical impression was a basal cell carcinoma.
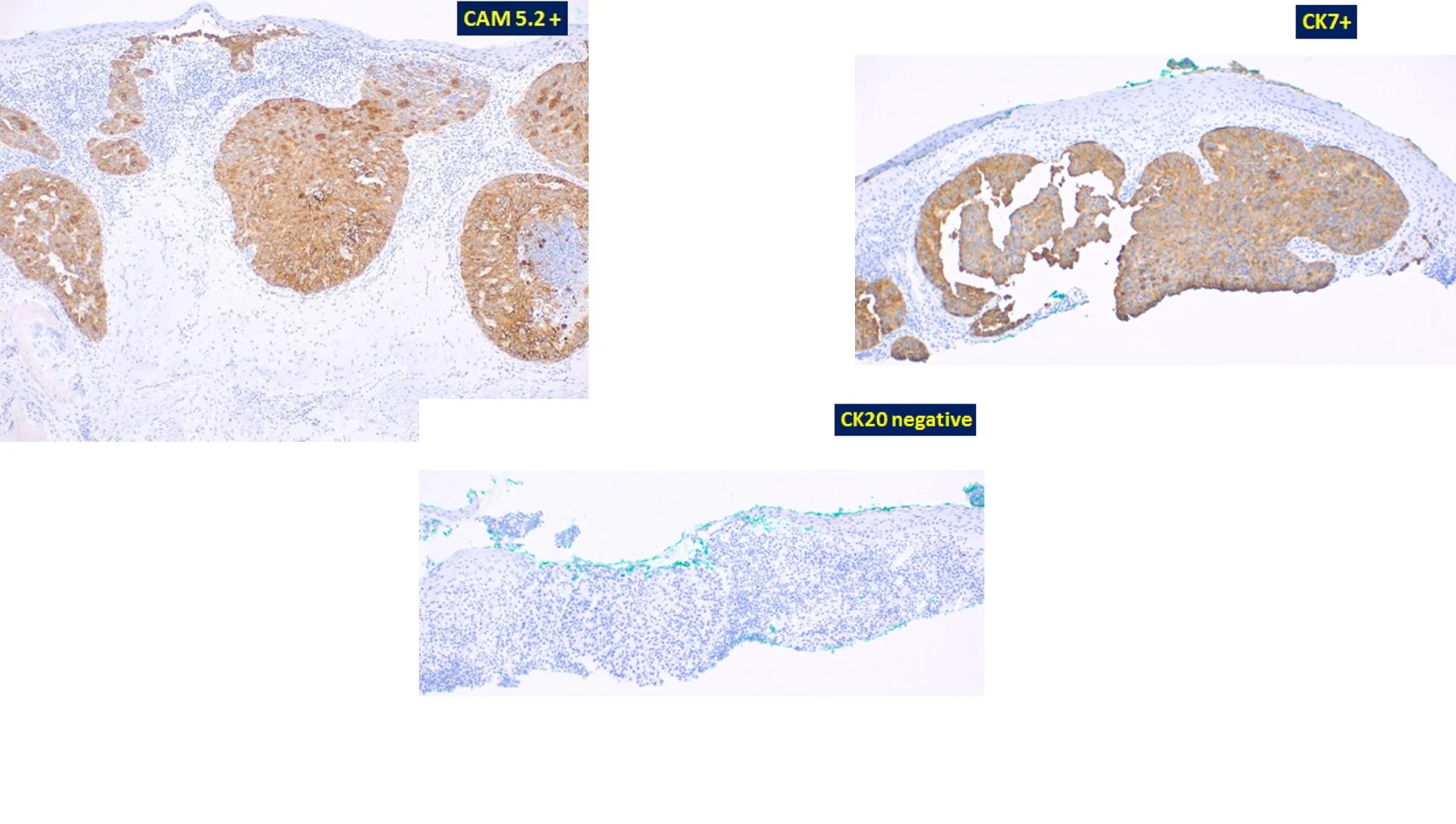
Histopathology: An initial shave biopsy from the lesion showed a poorly differentiated carcinoma located in the dermis, exhibiting focal contiguity with the overlying epidermis. The histology and immunophenotype of the tumor (p63- and CK7+) were most consistent with an adenocarcinoma, either primary cutaneous or an epidermotropic metastasis of a visceral carcinoma. As only a portion of the lesion was available for evaluation, complete excision and correlation with the residuum in the re-excision was recommended for a definitive diagnosis. Subsequent excision of the neoplasm showed a broad, poorly circumscribed proliferation of epithelial cells with abundant pale, eosinophilic cytoplasm and enlarged nuclei partially replacing the epidermis and involving some follicular infundibular units. Pagetoid scatter of the tumor cells was also noted. Aggregates of similar appearing cells were present in the superficial dermis. The neoplasm on repeat staining was confirmed to be p63 negative and Cytokeratin7, EMA and CAM 5.2 positive. A concomitant review of the patient’s original gastric tumor, showed distinct histologic features with columnar cells and hyperchromatic nuclei, unlike most of the cells in the current facial lesion. The cheek tumor was favored to represent “Extramammary Paget’s disease”, possibly an adenocarcinoma arising in Bowen’s disease. One could consider a metastatic epidermotropic adenocarcinoma, but the breadth, poor circumscription and very extensive intrafollicular component were considered unlikely histologic presentations of a visceral metastasis to the skin.
The lesion was adequately excised and the patient reports no subsequent recurrences over a period of 5 months.
Points of emphasis: Extramammary Paget's disease (EMPD) is a rare neoplasm mostly affecting apocrine gland bearing skin[1]. Although primarily seen in the anogenital area, the tumor can also rarely appear in non-apocrine bearing skin and is referred to as “ectopic extramammary Paget's disease” [2]. It is important for dermatopathologists to be aware of this rare variant of EMPD as it can histologically mimic primary cutaneous malignancies, especially Bowens disease and melanoma. Adequate immunohistochemical analysis aids in its diagnosis. The diagnosis also necessitates the need for a complete clinical work-up to exclude an underlying visceral adenocarcinoma. Primary cutaneous ectopic extramammary Paget's disease can be adequately treated with excision, including Mohs micrographic surgery and is associated with a good overall outcome and prognosis[3].
References: 1. Sarah H. Mehrtens, M.B., Ch.B., M.B. Sriramulu Tharakaram, and M.D. B.S., Extramammary Paget's Disease The New England Journal of Medicine, 2017. 376(17). 2. Hagiwara-Takita, A., et al., RANKL-Expressing Ectopic Extramammary Paget's Disease on the Lower Abdomen. Case Rep Dermatol, 2016. 8(2): p. 130-5. 3. Kim, S.J., et al., Surgical Treatment and Outcomes of Patients With Extramammary Paget Disease: A Cohort Study. Dermatol Surg, 2017. 43(5): p. 708-714.








Case 8: Onychocytic Matricoma
Caregivers: Jacqueline Brogan, MD, Jenny Yeh, MD, Morgan Fletcher, BS, Thomas Davis, MD; San Antonio, TX
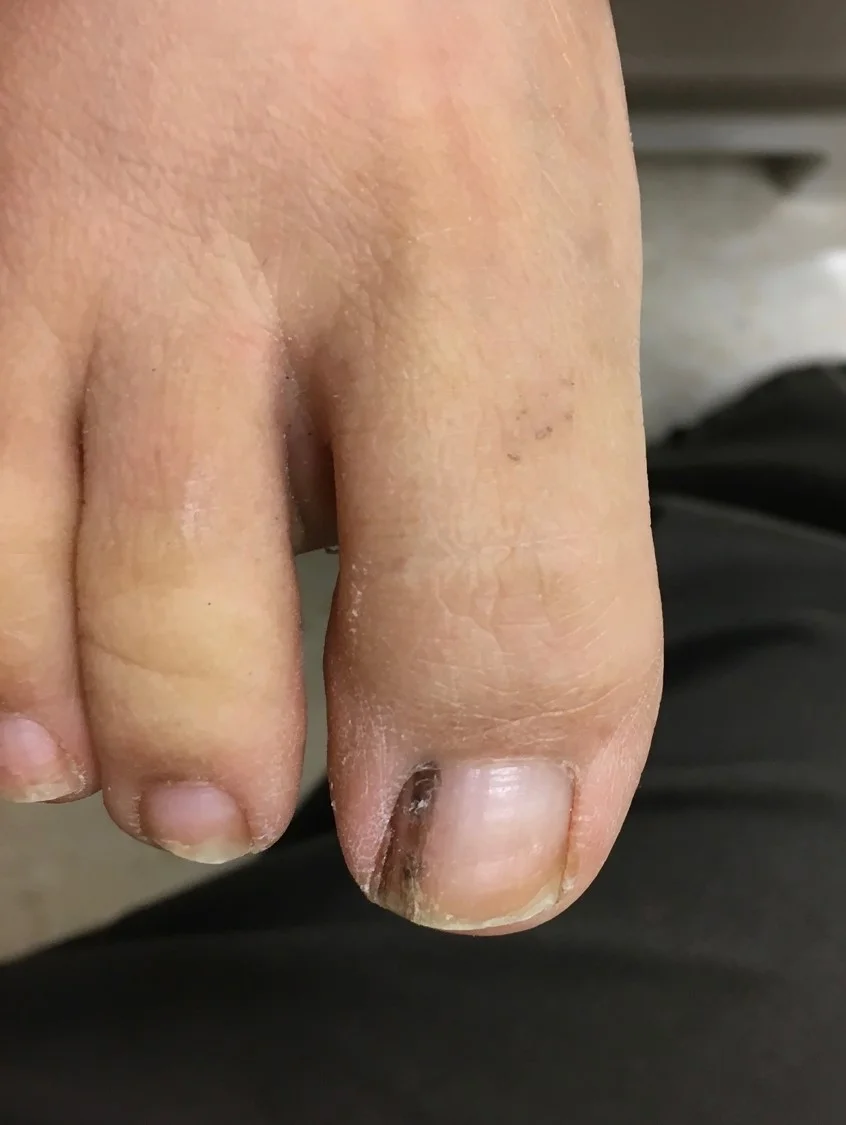
History: A 30 year-old female presented with a 3-month history of a dark streak appearing on her right first toenail. The lesion was asymptomatic and she had no other nail pigmentation.
Physical Examination: Physical exam revealed a 3.0mm wide, somewhat streaky, longitudinal brown streak associated with thickening of the nail.
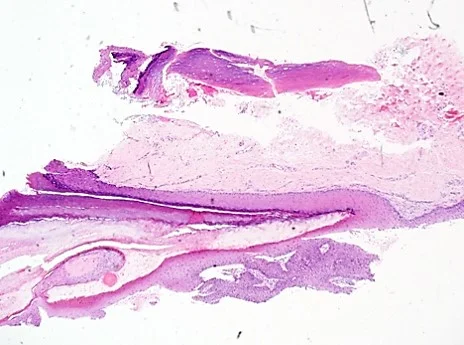
Histopathology: A 4.0mm punch biopsy of the nail matrix demonstrated a proliferation of the nail matrix epithelium with pigmented cells; MART-1 staining was negative.
Clinical Course: The patient did not return for follow up and was unable to be contacted to assure resolution of the lesion.
Diagnosis: Onychocytic matricoma
Points of Emphasis: Onychocytic matricoma (OCM) is a rare, benign, acanthoma of the nail matrix producing onychocytes. It is associated with acquired, localized thickening of the nail plate with longitudinal ridging and transverse hypercurvature, as well as longitudinal melanonychia (pachymelanonychia).1 On histopathology, the defining characteristics include endokeratinization in deeper layers of the tumor as well as prekeratogenous and keratogenous cells arranged in concentric nests.1 There may also be whorls of nail plate within the tumor stroma.2 This tumor can be classified according to pigmentation (pigmented or non-pigmented) and histological subtype (acanthotic, papillomatous or keratogenous with retarded maturation).1 Although pigmentation is often clinically present, melanocytes are typically not increased in number. 2 Our case can be classified as pigmented and acanthotic.
The differential diagnosis of OCM includes onychomatricoma (OM), onychocytic carcinoma (OCC), subungual keratoacanthoma, onychopapilloma and subungual melanoma. OM is a benign fibroepithelial matrical tumor that may also present as longitudinal pachyonychia with a thickened nail plate exhibiting ridging and transverse hypercurvature.3 The main histological factor that differentiates OM from OCM is the presence of prominent fibrous stroma with fibroblast proliferation in OM but not in OCM.3 As well, nail clippings can distinguish OM from OCM; while both exhibit thickened nail plates with cavities in a honeycomb pattern, OM nail clippings are characterized by large cavities, while OCM nail clippings are characterized by small cavities.3,4 OCC is a malignant epithelial matrical tumor that may also exhibit a similar clinical appearance as OCM.4,5 In contrast to OCM, OCC immunohistochemistry exhibits positive staining of p16 in the absence of p53 staining, indicative of an HPV-associated carcinoma.4 OCM can be distinguished from subungual keratoacanthoma and onychopapilloma through histopathologic findings.1,6
The most concerning differential of longitudinal melanonychia, often prompting nail biopsy, is subungual melanoma. Histopathology and immunochemistry distinguish subungual melanoma from OCM, as subungual melanoma classically displays cytologic atypia, pagetoid spread, confluent and multinucleated melanocytes, and lichenoid inflammatory reaction on histopathology; furthermore, immunoperoxidase stains (including HMB45, Melan-A, Sox-10 and MiTF) highlight the melanocytic proliferation in the matrix.7
As elucidated above, hematoxylin-eosin staining and immunohistochemistry of tissue samples is important in the diagnosis of OCM.1,3 Treatment recommendations for OCM have not been published, though excisional biopsy has been performed with no recurrence or persistence of the lesion.1,3
References: 1. Perrin C, Cannata GE, Bossard C, Grill JM, Ambrossetti D, Michiels JF. Onychocytic matricoma presenting as pachymelanonychia longitudinal: a new entity (report of five cases). Am J Dermatopathol. 2012;34(1):54-59. 2. Spaccarelli N1, Wanat KA, Miller CJ, Rubin AI. Hypopigmented onychocytic matricoma as a clinical mimic of onychomatricoma: clinical, intraoperative and histopathologic correlations. J Cutan Pathol. 2013;40(6):591-4. 3. Perrin C, Cannata GE, Langbein L, Ambrosetti D, Coutts M, Balaquer T, Garzon JM, Michiels JF. Acquired Localized Longitudinal Pachyonychia and Onychomatrical Tumors: A Comparative Study to Onychomatricomas (5 Cases)


Case 9: Acquired Subungual Fibrokeratoma in Pregnancy
Caregivers: Roya Nazarian, BA, Nikki Vyas, MD, Dana Stern, MD, Matthew Goldberg, MD, New York, NY
History: A 36-year-old female with no past medical history presented with a new onset nail lesion. She was in her 3rd trimester of pregnancy when she noted a new growth under the nail of her left first digit that was present for 3-4 weeks prior to presentation. The lesion occasionally bled, but she denied pain, pruritis, or other symptoms. She had no personal or family history of skin cancer.
Physical Examination: Physical exam was notable for a subungual, pink papule with a stalk that was 3-4 mm in length. All other exam findings were in normal limits.
Laboratory Data: Non-contributory.
Histopathology: An excisional biopsy was performed. Histopathologic exam of the lesion showed a fragment of nail plate and nail bed epithelium showing a flattened papule with overlying hyperkeratosis and fibrous stalk beneath the epidermis. The core of the papule showed dilated dermal vessels (oriented parallel along the vertical axis of the lesion) and increased, irregular collagen bundles, mimicking an acquired digital fibrokeratoma.
Diagnosis: The clinical and histopathological findings were most consistent with a benign subungual fibrokeratoma.
Clinical Course: The excisional site healed well without complication and the lesion did not recur.
Points of Emphasis: Acquired ungual fibrokeratoma is a rare and benign fibrous tissue tumor found in the periungual area. The term was first used in 1977 by Cahn et al, who believed the ungual fibrokeratoma was the same entity as the “garlic clove fibroma” first described in 1967.1 Because these tumors are exceedingly rare, they have not been clinically described well, yet a recent study of 20 patients found that 80% of ungual fibrokeratoms occur on toenails versus only 20% on the fingernails, and when found on the fingernails, only 5% are subungual.2 Lesions can present as rod-shaped, dome-shaped, flat, and branching. Histologically, acanthosis and thick collagen bundles oriented in the vertical axis with fibroblasts located in between collagen bundles can be seen.3 In 1985, Yasuki et al classified periungual fibrokeratoms into subtypes (Ip: within the proximal nail fold, Im: within the dermis beneath the nail matrix, Ib: within the nail bed, IIp: within the dermis of the dorsum of the distal phalanx, and IIl: within the lateral nail fold) based on the anatomical location.4 The case presented here would be classified as 1b because the lesion arises from the proximal nail folds in the subungual location with shallow, longitudinal, gutter-shaped depressions between two parallel ridges of the nail plate. While the exact etiology is unknown, both trauma and infectious etiology, mainly staphylococcal paronychia, have been proposed as predisposing factors.5,6 Treatment generally consists of complete surgical excision, as superficial excision has been noted to result in recurrence.7 We believe this is the first ever case describing an acquired periungual fibrokeratoma that developed during pregnancy, and we hope it will contribute to the existing literature by describing a novel presentation of a rare disease entity.
References: 1. Cahn RL. Acquired periungual fibrokeratoma. A rare benign tumor previously described as the garlic-clove fibroma. Archives of dermatology. 1977;113(11):1564-1568. 2. Hwang S, Kim M, Cho BK, Park HJ. Clinical characteristics of acquired ungual fibrokeratoma. Indian journal of dermatology, venereology and leprology. 2017;83(3):337-343. 3. Goktay F, Altan ZM, Haras ZB, et al. Multibranched acquired periungual fibrokeratomas with confounding histopathologic findings resembling papillomavirus infection: a report of two cases. Journal of cutaneous pathology. 2015;42(9):652-656. 4. Yasuki Y. Acquired periungual fibrokeratoma - A proposal for classification of periungual fibrous lesions. . The Journal of dermatology. 1985 12: 349-356. 5. Kint A, Baran R, De Keyser H. Acquired (digital) fibrokeratoma. Journal of the American Academy of Dermatology. 1985;12(5 Pt 1):816-821. 6. Sezer E, Bridges AG, Koseoglu D, Yuksek J. Acquired periungual fibrokeratoma developing after acute staphylococcal paronychia. European journal of dermatology : EJD. 2009;19(6):636-637. 7. Shelley WB, Phillips E. Recurring accessory "fingernail": periungual fibrokeratoma. Cutis. 1985;35(5):451-454.




Case 10: Basal Cell Carcinomas Appearing During Immunotherapy for Reported Metastatic Basal Cell Carcinoma
Caregivers: Philip R. Cohen, Shumei Kato, Aaron M. Goodman, Sadakatsu Ikeda and Razelle Kurzrock; National City, CA; La Jolla, CA; and Tokyo, Japan
History: A 58-year-old man presented with newly appearing scaly red plaques on his left shoulder and left clavicle. His past medical history was significant for not only multiple primary cutaneous basal cell carcinomas (BCCs), but also metastatic BCC to the brain, bone, liver and lungs 2 years earlier. His metastatic BCC had previously been unsuccessfully treated with vismodegib, stereotactic radiosurgery (for brain metastases), cisplatin and paclitaxel, sonidegib combined with buparlisib (a pan-class I PIK3 inhibitor), and vismodegib and paclitaxel. Hybrid capture-based next generation sequencing (HCNGS) of the metastatic BCC in the liver demonstrated a tumor mutational burden (TMB) of 103 mutations per megabase (/Mb; >19 mutation/Mb = high TMB) and 19 genomic alterations including amplification of PD-L1, PD-L2 and JAK2; he was started on the checkpoint inhibitor nivolumab 240 mg intravenously every 2 weeks, with remarkable and rapid improvement of performance status and tumor shrinkage to near complete remission. The new skin lesions had appeared in the setting of near complete remission of his metastatic disease on continued nivolumab treatment.
Physical Examination: Erythematous plaques appeared on his left anterior shoulder and left chest.
Laboratory Data: HCNGS of the primary cutaneous skin tumors demonstrated a tumor mutational burden of 45 mutations/Mb and 8 genomic alterations. In contrast to the metastatic BCC in his liver, the primary skin cancers did not demonstrate amplification of PD-L1, PD-L2 or JAK2.
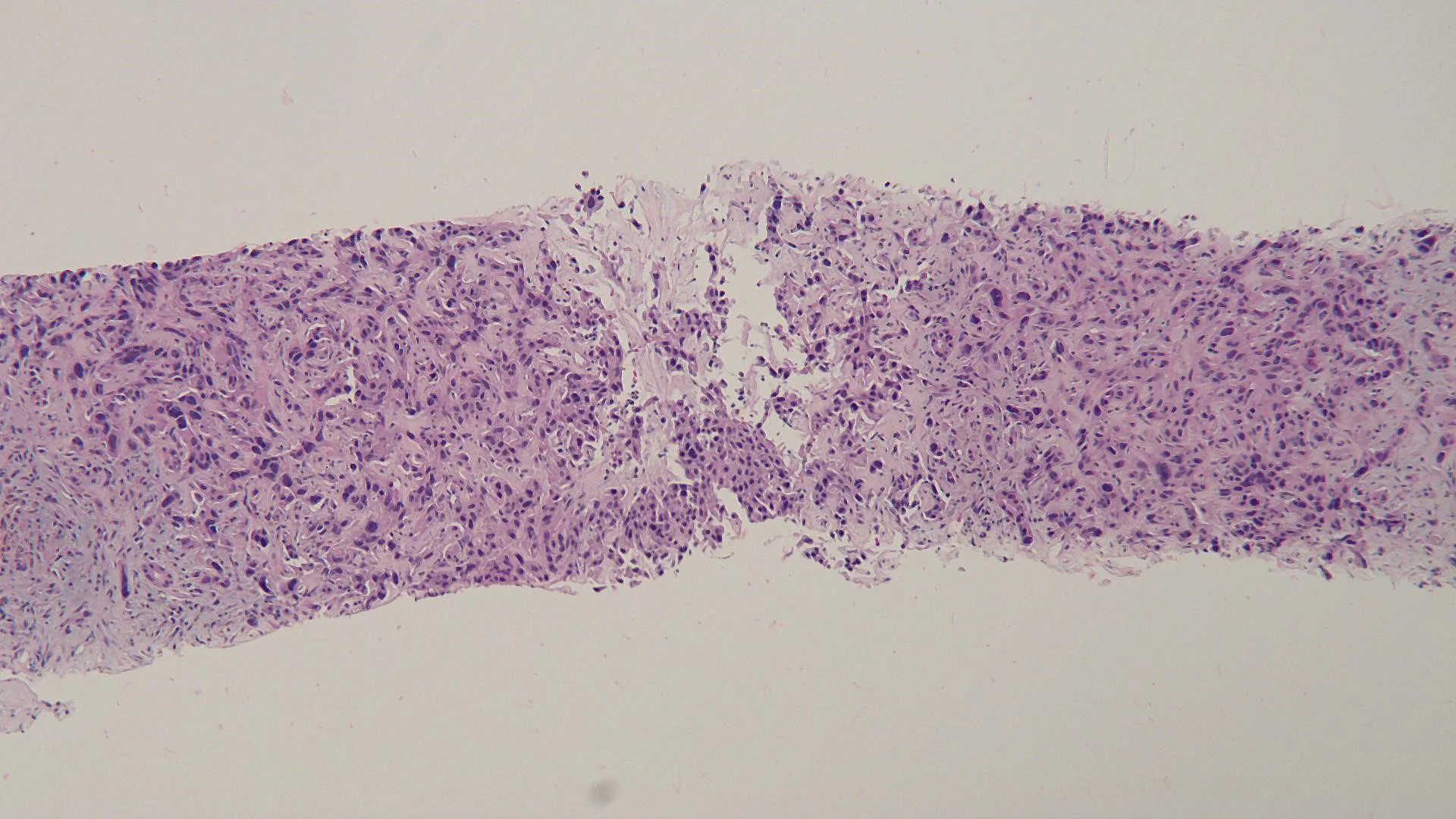
Histopathology: Superficial buds of basaloid tumor cells extend from the overlying epidermis into the dermis.
Clinical Course: The new skin BCCs were each treated with electrodessication and curettage. Follow up exam showed complete healing of the treated skin cancer sites without tumor recurrence. He continues to receive nivolumab every other week and his metastatic BCC continues to show over 95% regression on imaging.
Diagnosis: Basal cell carcinoma: new cutaneous tumors during successful immunotherapy for metastatic disease.
Points of Emphasis: Genomically targeted therapies aimed at the Hedgehog pathway, such as vismodegib and sonidegib, are effective agents for treating patients with metastatic BCC. However, the patient’s metastatic BCC was resistant to these targeted therapies; indeed, his lack of response to treatments targeting a single genomic aberration is not surprising since his metastatic liver tumor had numerous alterations [1,2]. Checkpoint inhibitors, such as nivolumab, may be effective immunotherapy agents for patients whose tumors have increased PD-L1 expression or multiple genomic aberrations (increased TMB) or both [3]. Of interest was the observation that in the setting of ‘late’ metastatic disease, the patient’s metastatic BCC was highly susceptible to anti-PD1 immunotherapy. Yet, he continued to develop new primary cutaneous BCCs--even though he was receiving nivolumab and his metastatic disease remained in near complete remission. The checkpoint inhibitor did not prevent his new skin cancers, perhaps since the ‘early’ primary disease had a lower TMB. In summary, immunotherapy may be best suited for ‘late’ disease in which the metastatic tumor has many genomic aberrations and an increased TMB in contrast to ‘early’ disease in which the primary cancer has fewer molecular alterations and a lower TMB [1].
References: 1. Cohen PR, Kato S, Goodman AM, Ikeda S, Kurzrock R. Appearance of new cutaneous superficial basal cell carcinomas during successful nivolumab treatment of refractory metastatic disease: implications for immunotherapy in early versus late disease. Int J Mol Sci 2017;18(8). pii: E1663. doi: 10.3390/ijms18081663 . 2. Ikeda S, Goodman AM, Cohen PR, Jensen TJ, Ellison CK, Frampton G, Miller V, Patel SP, Kurzrock R. Metastatic basal cell carcinoma with amplification of PD-L1: exceptional response to anti-PD1 therapy. NPJ Genom Med 2016;1. pii: 16037. doi: 10.1038/npjgenmed.2016.37 . 3. Goodman AM, Kato S, Cohen PR, Boichard A, Frampton G, Miller V, Stephens, PJ, Daniels GA, Kurzrock R. Genomic landscape of advanced basal cell carcinoma: Implications for precision treatment with targeted and immune therapies. Oncoimmunology 2017;7(3):e1404217. doi: 10.1080/2162402X.2017.1404217


Case 11: Cutaneous Metastatic Sarcomatoid Renal Cell Carcinoma and Hand-Foot Syndrome
Caregivers: Sarah Oberhelman, B.S., Emily Powell, M.D., Pamela Martin, M.D., Andrea Murina, M.D.; New Orleans, LA
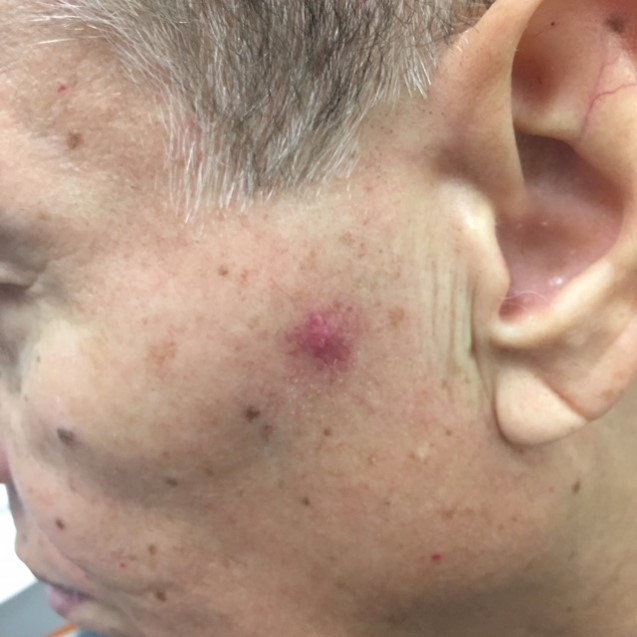
History: A 58-year-old Hispanic man with stage 4 renal cell carcinoma (RCC) was referred to dermatology by hematology/oncology for evaluation of a palmar rash. The patient reported starting pazopanib 3 months prior following the diagnosis of metastatic RCC of the left femur and rib. He had stopped the medication for a month interval during which time he underwent left femur curettage with subsequent adjuvant radiation treatment to the femur. He had since restarted pazopanib. His palmar rash developed 1.5 months before presentation as painful red, hyperkeratotic papules and patches on his palms that enlarged and blistered. The lesions would eventually callous and fall off. During his initial evaluation, an unrelated red papule on the left cheek was noted during examination. The lesion had been present for the previous 2-3 months and was mildly tender.
Physical Examination: The patient was pleasant and in no acute distress. A single 2-3 mm mildly tender erythematous papule with surrounding telangiectasias was present on the left cheek. There was no palpable regional lymphadenopathy. Numerous erythematous and scaly papules in various stages of development were present on the bilateral palms and elbows.
Histopathology: Punch biopsies of the left cheek and left elbow were performed. The left cheek biopsy showed a malignant spindle and epithelioid cell neoplasm. Immunohistochemical staining was positive for epithelial membrane antigen, pan-cytokeratin, vimentin, CD10, and PAX8 and negative for RCC antigen and PAX2, consistent with cutaneous, metastatic sarcomatoid renal cell carcinoma. The left elbow biopsy showed psoriasiform chronic dermatitis with eosinophilia.
Clinical Course: Following review of the histopathology from the left cheek biopsy, the patient was seen in clinic for suture removal and biopsy results were discussed with the patient. The patient was referred back to hematology/oncology where he subsequently received palliative radiation treatment. The palmar and elbow lesions were concerning for hand-foot syndrome, also known as palmoplantar erythrodysesthesia, given the patient’s history of a desquamating dermatitis following pazopanib treatment, physical exam findings, and consistent histopathology.1 The lesions improved with Clobetasol 0.05% cream applied twice daily.
Diagnosis: Cutaneous Metastatic Sarcomatoid Renal Cell Carcinoma and Hand-Foot Syndrome
Points of Emphasis: Cutaneous metastasis is a rare complication of any malignancy, most commonly seen in breast, lung, colon, and ovarian carcinomas as well as malignant melanoma.2 RCC metastasis only accounts for 4.6% of cutaneous metastases, most commonly occurring in patients with known clear cell carcinoma.3 Sarcomatoid differentiation is a form of high-grade transformation that can be seen in any histologic subtype of RCC and accounts for 5% of all primary RCC.4 Histologically, metastatic sarcomatoid RCC tumors appear similar to primary cutaneous tumors, so a high index of suspicion and immunohistochemical staining are essential for diagnosis.4 Vimentin and cytokeratin positivity are together suggestive of metastatic RCC rather than a primary tumor.5 While there is currently no good marker of sarcomatoid RCC available, PAX8 is considered the most sensitive marker, labeling 0-50% of metastatic lesions.4,5 Notably, PAX2 and RCC antigen are only expressed in up to 22% of primary sarcomatoid RCC, and metastatic sarcomatoid RCC is historically negative for PAX2.5
References: 1. Que Y, Liang Y, Zhao J, Ding Y, Peng R, Guan Y, Zhang X. Treatment-related adverse effects with pazopanib, sorafenib and sunitinib in patients with advanced soft tissue sarcoma: a pooled analysis. Cancer Manag Res. 2018;10:2141-2150. 2. Kishore M, Chauhan DS, Dogra S. Unusual presentation of renal cell carcinoma: A rare case report. J Lab Physicians. 2018;10(2):241-244. 3. Plaza JA, Perez-Montiel D, Mayerson J, Morrison C, Suster S. Metastases of soft tissue: a review of 118 cases over a 30-year period. Cancer. 2008;112:193-203. 4. Logunova V, Sokumbi O, Iczkowski KA. Metastatic sarcomatoid renal cell carcinoma manifesting as a subcutaneous soft tissue mass. Journal of Cutaneous Pathology. 2017;44:874-877. 5. Truong LD, Shen SS. Immunohistochemical diagnosis of renal neoplasms. Arch Pathol Lab Med. 2011; 135:92-108.









Case 12: Merkel Cell Carcinoma
Caregivers: Christopher M. Lowther, MD, Troy Fiddler MD, PhD, Juanita Sapp, MD, Clay J. Cockerell, MD; Cody, WY, Billings, MT, Powell, WY, and Dallas, Texas
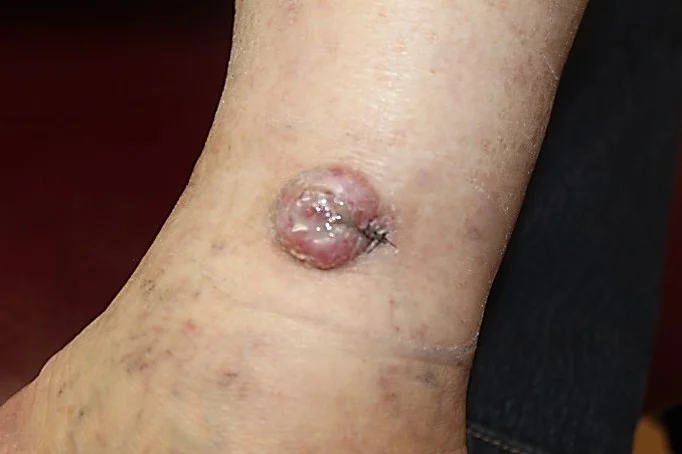
History: The 61-year-old white female presented with the four month history of a non-healing ulcerated mass approximately 3.5 cm in diameter on her right ankle.
Physical Examination: The patient had two small lesions, both on her right lower leg measuring 5 mm in diameter. Her ankle was painful and inflamed. She had diarrhea which started after two courses of doxycycline antibiotics, prior to referral. Lymphadenopathy was noted in the right inguinal area. The rest of her physical exam was unremarkable. All three lesions on her right leg underwent biopsy.
Laboratory Data: The only abnormal laboratory findings were hemoglobin was 10.9 g/dL with an MCV of 86.5 fl. A serum ferritin was 9 ng/ML (eight - 252 ng/ML). The consultant initially diagnosed metastatic squamous cell carcinoma versus pyoderma gangrenosum. The patient had no history of prior inflammatory bowel disease although there was a strong family history for colon cancer.
Histopathology: All three lesions were biopsied and demonstrated malignant basophilic neuroendocrine neoplasms thought to be either primary or metastatic. Immunoperoxidase stains were strongly positive for cytokeratin 20 and synaptophysin and negative for cytokeratin cocktail and TTF-1. A stain for CEA highlighted some of the background leukocytes. The diagnosis of neuroendocrine carcinoma, likely Merkel cell carcinoma possibly metastatic, was rendered.
Clinical course: Computerized axial tomography revealed small non-calcified pulmonary nodules the largest measuring 6 mm in greatest diameter. There was a small 7 mm abnormality in the liver. There were large and suspicious appearing right-sided inguinal and lower abdominal wall lymph nodes in the right inguinal region and right iliac region. PET imaging was performed, from the skull to the toes. Focal hypermetabolic activity was noted at the left base of the tongue, with no hypermetabolic activity in the lungs. There was no evidence of mediastinal or hilar lymphadenopathy.
Liver, gallbladder, spleen, pancreas, adrenal glands, kidney and vessels were unremarkable. The gastrointestinal track is unremarkable. Right inguinal lymphadenopathy with the largest lymph node measuring 19 x 21 mm with hypermetabolic activity maximum SUV value of 4.1 suspicious for metastasis.
Lymph nodes were seen along the right mid-thigh and lower leg medial with hyper metabolic activity. Diffuse soft tissue thickening around the distal tibia and fibula around the ankle with soft tissue defect and hypermetabolic activity maximum SUV value at 9.4 at the lateral ankle suspicious for known carcinoma. Biopsy was subsequently performed on the right inguinal lymph node and consistent with Merkel cell carcinoma.
Diagnosis: Metastatic Merkel Cell Carcinoma, StagingT2, N3, MO equating to stage IIIB. The case was presented at St. Vincent healthcare tumor board and recommendation was to proceed with systemic therapy due to extent of disease. StagingT2,N3, MO equating to stage IIIB.
The newly approved PD-LI for Merkel cell carcinoma, Avelumab, was started. She was not considered a surgical candidate by the tumor board. Following the start of cycle four of chemotherapy, the right ankle had almost completely resolved. She tolerated the treatment without complications. Follow-up axial tomography of chest abdomen and pelvis and right lower leg was ordered.
Points of emphasis: The primary lesion of Merkel cell carcinoma is often distinguished by the absence of distinctive clinical characteristics and is rarely suspected at the time of biopsy. It often presents as a rapidly growing reddish blue dermal papule or nodule.5
Merkel cell carcinoma of the skin was first described by Toker in 1972 and initially thought by him to represent a sweat gland carcinoma. Further study revealed the presence of dense corps granules of the tactile Merkel cells.1,2,3,4 Upon presentation 66% of patients have local disease, 27% nodal involvement and 7% distal metastasis.2,4 The majority of patients with Merkel cell carcinoma are 70 years or older with an increased incidence in renal transplant patients, chronic lymphocytic leukemia and HIV.1 Polyomavirus is associated with 80% of tumors.4 The primary lesion of Merkel cell carcinoma is often distinguished by the absence of distinctive clinical characteristics and is rarely suspected at the time of biopsy. It often presents as a rapidly growing reddish blue dermal papule or nodule.5
Avelumab, the human anti-PD-L1 antibody, is the first immunotherapy approved for metastatic Merkel cell carcinoma, for adult and pediatric patients 12 years and older with metastatic Merkel cell carcinoma. Metastatic Merkel cell carcinoma is a rare and aggressive skin cancer with fewer than half the patients surviving more than one year and fewer than 20% surviving beyond five years.6 Immunochemistry is often definitive. Merkel cells are almost impossible to see on light microscopy in normal skin and without immunohistochemical stains are nearly impossible to detect.5 CK 20 is the predominant tool used by pathologists and stained approximately 80 to 90% of all Merkel cell carcinomas and a distinctive para nuclear-dot like pattern.5,7
Approximately 53% of Merkel cell carcinomas occur in the head and neck and 35% occur on the extremities. In the Javelin Merkel Cell Carcinoma 200, a phase 2 clinical trial, at the median follow-up of 16.4 months of which 88 patients had been enrolled and treated with Avelumab, the overall response rate was 33% (29 patients), partial response rate was 22% and complete response was 11%. 86% of tumor responses lasted at least six months (25 patients) and 45% lasted at least 12 months (13 patients).8
References: 1.Toker C. Trabecular carcinoma of the skin. Arch of Dermatol 1972; 105:107 – 110 2. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Amer. 2012; 26:1351 – 1374 3. Goessling, W, Mckee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002; 20:588 – 598. 4. Oram C , Bartus C S, Purcell S. Merkel cell carcinoma: A review. Cutis. 2016 Volume 97, 4 to 90 – 295 5. Wang T, Byrne, P, Et al, Semin Cutan Med Surg. 2011 Mar:30 (1) 48-56 . 6. Lemos B, Storer B, Lyer J, et al. Pathologic nodal evaluation approves prognostic accuracy and Merkel cell carcinoma; J Amer Acad Derm. 2010; 63(5): 751 – 761. 7. Bobos M, Hytiroglou P, Kostopoulos I, et al. immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Dermatopathol. 2006; 28:99 – 104. (Pub Med) 8. Pearson, J , Meyers, A Skin cancer – Merkel cell carcinoma, Medscape updated March 27, 2017





Case 13: Ewing’s Sarcoma in an Adult- A Rare Entity
Caregivers: Leah Persad, DO, Amit Patel, MD, Jerad Gardner, MD Clay Cockerell, MD; Dallas, TX, Nederland, TX, Little Rock, AR
History: A 75-year-old woman presented with a 1 cm erythematous nodule on the right anterior shoulder. The lesion had been present for an undetermined period of time.
Physical Exam: Involving the right anterior shoulder was a 1 cm, round pink to erythematous smooth firm nodule.
Histopathology: A biopsy of the lesion revealed dermal sheets of small round blue cells with uniform round nuclei, scant cytoplasm, relatively minimal pleomorphism but with many mitotic figures. The tumor was diffusely and strongly positive for CD99, showed rare positivity for synaptophysin and was negative for EMA, Pan-CK, CD45, SOX-10, SMA, CK20, TTF-1, CD43, MUM-1, TdT, CD34, and p40. Cytogenetic and molecular testing of the tumor was positive for rearrangement of the EWSR1 (22q12) locus by FISH. An EWSR1/FLI1 fusion transcript was detected by RT-DNA amplification respectively.
Clinical Course: The patient was referred to MD Anderson Cancer Center for further workup to rule out additional involvement.
Diagnosis: Primary Cutaneous Ewing Sarcoma in an Adult
Points of Emphasis: Ewing sarcoma (ES) is a small round blue cell tumor that is closely related to the primitive neuroectodermal tumor (PNET) family. Ewing sarcoma is typically a neoplasm that occurs in bone or soft tissue of children and young adults. Rarely, it can present as a primary cutaneous neoplasm in adults. In this setting, the mean age is 22 years (range from 22 months to 77 years) with a nearly 2:1 female predominance. The median size of tumors reported was 2-3 cm most commonly on the extremities, followed by the head and trunk. Radiologic and clinical correlation is essential to confirm that the lesion is small and confined to the skin rather than a metastasis or direct extension from deep soft tissue or bone.
Histopathology typically reveals a nodular or sheet-like proliferation of undifferentiated small round blue cells. In this setting, there is a wide range of differential diagnoses when occurring in an adult including Merkel cell carcinoma, metastatic small cell carcinoma of the lung, lymphoma and small cell melanoma. Ancillary studies can help further differentiate these entities. ES/PNET almost always characteristically show diffuse positivity for CD99 in a membranous pattern. Rarely, Merkel cell carcinomas and lymphoblastic lymphoma can express CD99 so that other stains may be required to distinguish these, however. ES/PNET may infrequently display focal positivity for pancytokeratin, S100, and neuroendocrine markers. Based on the varied immunohistochemical pattern of staining, there can be some diagnostic confusion between these entities, in which morphological and clinical correlation can aid in the diagnosis.
The characteristic chromosomal translocation in ES/PNET t(11;22)(q24; q12) resulting in the EWSR1-FLI1 fusion gene can greatly aid in the diagnosis. However, approximately 10% of ES/PNET have variant translocations. Rarely, the EWSR1 gene can be identified in other morphologically distinct entities as it has been described as a “promiscuous” gene.
Dual color break-apart EWS FISH probes can detect EWSR1 rearrangements regardless of the translocation variants. In this break-apart probe strategy, fluorosceinated probes normally flank the EWSR1 gene. In the nucleus, abnormal (separate) red and green fluorochromes signify disruption of one copy of the EWSR1 gene. In contrast, the normal allele is intact as evidenced by fused red and green signals indicating that the two probes are juxtaposed as a consequence of binding the same chromosomal locus.
Based on the relatively limited data in the literature on primary cutaneous Ewing sarcoma, it appears to have a much better prognosis than its skeletal or deep soft tissue counterpart. It has been established that the 10-year probability of survival was 91% with rare chance of metastatic disease. In light of the excellent prognosis of this tumor when confined to the skin, some authors have suggested that surgical excision should be the primary mode of treatment with adjuvant therapy such as chemotherapy or radiotherapy playing a lesser role.
References: 1. Delaplace M, Lhommet C, de Pinieux G, Vergier B, de Muret A, Machet L. Primary cutaneous Ewing sarcoma: a systematic review focused on treatment and outcome. Br J Dermatol. 2012 Apr;166(4):721-6. doi: 10.1111/j.1365-2133.2011.10743.x. Epub 2012 Mar 5. 2. Boland JM, Folpe AL. Cutaneous neoplasms showing EWSR1 rearrangement. Adv Anat Pathol. 2013 Mar;20(2):75-85. doi: 10.1097/PAP.0b013e31828625bf. 3. Machado I, Traves V, Cruz J, Llombart B, Navarro S, Llombart-Bosch A. Superficial small round-cell tumors with special reference to the Ewing's sarcoma family of tumors and the spectrum of differential diagnosis. Semin Diagn Pathol. 2013 Feb;30(1):85-94. doi: 10.1053/j.semdp.2012.01.007. 4. Shingde MV, Buckland M, Busam KJ, McCarthy SW, Wilmott J, Thompson JF, Scolyer RA. Primary cutaneous Ewing sarcoma/primitive neuroectodermal tumour: a clinicopathological analysis of seven cases highlighting diagnostic pitfalls and the role of FISH testing in diagnosis. J Clin Pathol. 2009 Oct;62(10):915-9 5. Collier AB 3rd, Simpson L, Monteleone P. Cutaneous Ewing sarcoma: report of 2 cases and literature review of presentation, treatment, and outcome of 76 other reported cases. J Pediatr Hematol Oncol. 2011 Dec;33(8):631-4







Case 14: Acquired Unilateral Nevoid Telangiectasia
Caregivers: Julia Accetta, MD, Cather McKay MD, and Howard Patrick Ragland MD; New Orleans, LA
History: A 60-year-old female with a history of rheumatoid arthritis and Sjogren's syndrome well-controlled on tocilizumab, methotrexate, and hydroxychloroquine, presented for evaluation of an asymptomatic red lesion on the plantar surface of her right foot. Her regular pedicurist brought the lesion to her attention at their last visit four weeks ago and since then, she has not noticed any changes. She denies associated pain or itching. No similar lesions elsewhere.
Physical Examination: Patient is well-appearing. On the right plantar foot there is a non-tender partially blanchable retiform deep-red patch extending from the great toe to the instep. She has no other skin lesions on total body skin exam. She has diffuse joint deformities including deviations of MCPs and metatarsals.
Laboratory Data: Recent complete blood count, comprehensive metabolic profile, erythrocyte sedimentation rate, and C-reactive protein were all within normal limits.
Histopathology: Punch biopsy of right plantar foot showed a proliferation of dilated blood vessels in the epidermis consistent with telangiectasia. There was no evidence of thromboembolism, malignancy, or vasculitis.
Clinical Course: Given the clinical and histopathologic findings, the patient was monitored and no change in the current lesion and no new lesions have appeared.
Diagnosis: Acquired unilateral nevoid telangiectasia
Points of Emphasis: Unilateral nevoid telangiectasia (UNT) is a vascular dermatosis characterized by superficial telangiectasias in a unilateral and segmental distribution. UNT can be either congenital or acquired. The congenital form is due to a rare, autosomal dominant somatic mutation that occurs most commonly in males shortly after the neonatal period. The acquired form is thought to be secondary from elevated estrogen. Although rarely reported in the literature, UNT may be common but under diagnosed due to its asymptomatic presentation.
Clinically, UNT appears as multiple patches of superficial, blanchable telangiectasias that appear in a unilateral, linear distribution. The lesions may be Blaschkoid or follow a dermatomal distribution, favoring the third and fourth cervical dermatomes. Sometimes, an anemic halo, or pale ring surrounding the telangiectasia may be noted. Differential includes hemangioma, primary telangiectases i.e. linear atrophoderma of Moulin, angioma serpiginosum and secondary telangiectases i.e. erythema ab Igne or an adverse effect from topical steroids (1).
On histology, there are multiple, dilated, thin-walled vessels in the superficial papillary dermis, without cell proliferation or angiogenesis and minimal inflammation (2). Laser-Doppler flowmetry can identify alterations in local blood flow such as hyperperfusion.
The pathogenesis remains relatively unknown, although it commonly presents in women with high estrogen states, such as puberty, pregnancy, and oral contraceptive use as well as individuals with alcoholic hepatitis, chronic liver disease, and hepatitis B and C infections (3). Rarely it is associated with liver metastasis from patients with colon cancer or carcinoid tumor (4,5). Hepatic failure is known to cause hyperestrogenemia that leads to vascular abnormalities such as spider angiomas.
Despite the association with estrogen, numerous studies of patients with UNT have confirmed normal estrogen levels as well as negative estrogen and progesterone receptors. In fact, all documented cases of increased serum estrogen were noted only in pregnant patients (6). Only one case report has identified elevated estrogen receptors in lesional skin (1). UNT may flare with pregnancy and resolve post-partum, but the topical application of estrogen cream in a previously affected woman did not induce recurrence of telangiectasias (7).
Other theories include chromosomal mosaicism, elevation of angiogenic factors such as VEGF, hemodynamic disturbances, neural alterations, and aberrations in connective tissue (1,9). Numerous other cases have been described without a known pathology, suggesting that the condition may not be as rare as previously described and is simply under diagnosed.
UNT is indolent and asymptomatic, with treatment catered more towards good aesthetic outcome using cosmetic camouflage. In some cases of modifiable hyperestrogenemia, removal may improve the disease. Alternatively, pulse dye lasers (wavelength 585 nm) provide moderate response, although recurrence may be common.
References: 1. Wenson SF, Jan F, Sepehr A. Unilateral nevoid telangiectasia syndrome: a case report and review of the literature. 2011; 17(5):2. 2. Hynes LR, Shenefelt PD. Unilateral nevoid telangiectasia: Occurrence in two patients with hepatitis C. J Am Acad Dermatol. 1997;36:819–22. 3. Guedes R, Leite L. Unilateral nevoid telangiectasia: a rare disease?. Indian J Dermatol. 2012; 57(2):138-140. 4. Beacham BE, Kurgansky D. Unilateral naevoid telangiectasia syndrome associated with metastatic carcinoid tumour. Br J Dermatol. 1991;124:86–88. 5. Kim HJ, Kim KO, Kim Y, et al. Acquired unilateral nevoid telangiectasia accompanied by liver metasis of colon cancer. Annal Dermatol. 2016; 28(3):404-405.




Case 15A: Stewart-Treves Syndrome at site of Cavernous Lymphangioma
Caregivers: Alexandra Steele, BS, Margaret Brown, MD, Valerie Shiu, MD, Sandra Osswald, MD; San Antonio, TX
History: A 48-year-old female with history of cavernous lymphangioma of the right posterior distal thigh, which was excised and re-excised several times during childhood and adulthood, presented to the orthopedic surgery clinic with recurrence of the mass. At that time, the mass was described as a 15 by 12 centimeter mobile mass on the posterior distal thigh. Initial excision by orthopedic surgery revealed lymphangiosarcoma with positive margins. She then underwent re-excision with negative margins and repair with skin grafting. Imaging with PET-CT showed no evidence of metastatic disease. She was treated with adjuvant radiation and was followed closely. Her course was complicated by lymphedema and poor wound healing of the right lower extremity requiring repeat skin grafting. Roughly two years later, she noticed several new nodules on her right shin. Shave biopsy of one of these nodules by an outside dermatologist was consistent with angiosarcoma. She was started on Paclitaxel, but this was discontinued in the setting of neuropathy. She was then started on Doxorubicin, which also had to be discontinued because of continued poor wound healing. She underwent a second course of radiation with improvement. Approximately one year later, and three years after her initial diagnosis of lymphangiosarcoma, she was referred to our dermatology clinic by her oncologist for a new tender bump adjacent to her surgical site.
Physical Examination: Right posterior distal thigh: 0.4 centimeter violaceous papule adjacent to an approximately 25 centimeter area of extensive scarring involving the popliteal fossa. The right lower leg is edematous when compared to the left.
Laboratory/Imaging Data: MRI, right leg: mildly increased size of an ill-defined soft tissue along the superiomedial margin of the surgical defect; soft tissue post-treatment edema throughout the thigh. CT chest, abdomen, pelvis: internal development subsegmental/subpleural reticular nodular opacities in the slightly more superior left lower lobe, favor atelectasis with underlying nodule not entirely excluded; no CT evidence of metastatic disease in the abdomen or pelvis.
Histopathology: Punch biopsy from violaceous papule revealed a diffusely infiltrative tumor with poorly formed vascular slits lined by pleomorphic nuclei dissecting through collagen bundles.
Clinical Course: The patient continues to follow closely with medical oncology and orthopedic surgery. Above the knee amputation versus hip disarticulation are currently being considered to achieve local control. Her lung nodules will continue to be monitored closely to survey for metastatic disease, but none has been identified at this time.
Diagnosis: Recurrent angiosarcoma, Stewart-Treves syndrome at site of cavernous lymphangioma
Points of Emphasis: Angiosarcomas are rare sarcomas of endothelial origin comprised of vascular or lymphatic proliferations. They can occur internally within visceral organs or cutaneously. Although extremely rare, accounting for 1%-2% of all sarcomas, their prognosis is particularly poor with a 5-year survival of approximately 30% for localized disease [1].
Cutaneous angiosarcomas (cAS) present as violaceous macules, patches, papules, plaques or nodules that can be ulcerative and multifocal. They are classified into three groups: primary sporadic, post-radiation and lymphedema-associated (referred to as Stewart-Treves syndrome). Primary sporadic cAS are the most common form and often appear in the sun-exposed head and neck region of elderly patients, with a predilection for white men [2]. Post-radiation cAS appear within or near a previously irradiated area. Lymphedema-associated cAS or Stewart-Treves syndrome presents in the setting of lymphedema. Although the overwhelming majority of these cAS occur after mastectomy with lymph node dissection, they can be a consequence of chronic lymphedema caused by any process including obesity, trauma, filariasis and even congenital lymphatic malformations [2,3].
The etiology of cAS is likely multifactorial, although mutations in tumor suppressor genes, such as TP53, as well as genes involved in the activation and suppression of angiogenesis (PTPRB and PLCG1) are thought to play a role [4]. The lymphedema-associated form is thought to be in part due to increased interstitial protein in areas of lymphostasis causing local lymphangiogenesis and angiogenesis [4].
Early clinical suspicion and careful histological evaluation are necessary for a favorable outcome. Histologic features consistent with cAS include infiltrative spaces, lined by pleomorphic endothelial cells, invading the dermal collagen. Significant hemorrhage and blood-filled spaces may be seen. Superficial biopsies of well-differentiated lesions may be misinterpreted as hemangioma or lymphangioma. Poorly differentiated cAS can appear similar to melanoma or carcinoma, with sheets of mitotically active, pleomorphic cells with ill-defined vascular structures [4]. On immunohistochemical staining, most cAS are positive for endothelial markers such as CD31, CD43, ERG and FLI1, although other lesions included in the usual differential are as well. Staining for c-MYC can be helpful in distinguishing secondary cAS from atypical vascular lesions in irradiated or edematous sites, although most primary sporadic forms do not demonstrate c-MYC amplification [5].
Due to the aggressive nature and poor prognosis of cAS, surgical excision with wide margins is the gold standard of treatment [2]. Even when clear surgical margins are attained, recurrence and metastasis are common. Therefore, adjuvant radiation to the excised site is recommended. In the setting of metastatic or locally advanced disease, palliative chemotherapy with agents such as doxorubicin or paclitaxel may be considered [2]. Most recently, treatment modalities targeting VEGF such as bevacizumab and tyrosine kinase inhibitors have been of interest but more research needs to be done to establish their efficacy [1].
References: 1. Dossett LA, Harrington M, Cruse CW, Gonzalez RJ. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39(4):258-263. 2. Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol. 2010;11(10):983-991. 3. Miyagawa, T. et al. Lymphangiosarcoma of the hip arising in a congenital non-irradiated lymphangioma. J Dtsch Dermatol Ges. 2017;15(12):1235-1237. 4. Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925. 5. Brenn, T. Angiosarcomas of the skin. Pathologe. 2015;36(1):70.

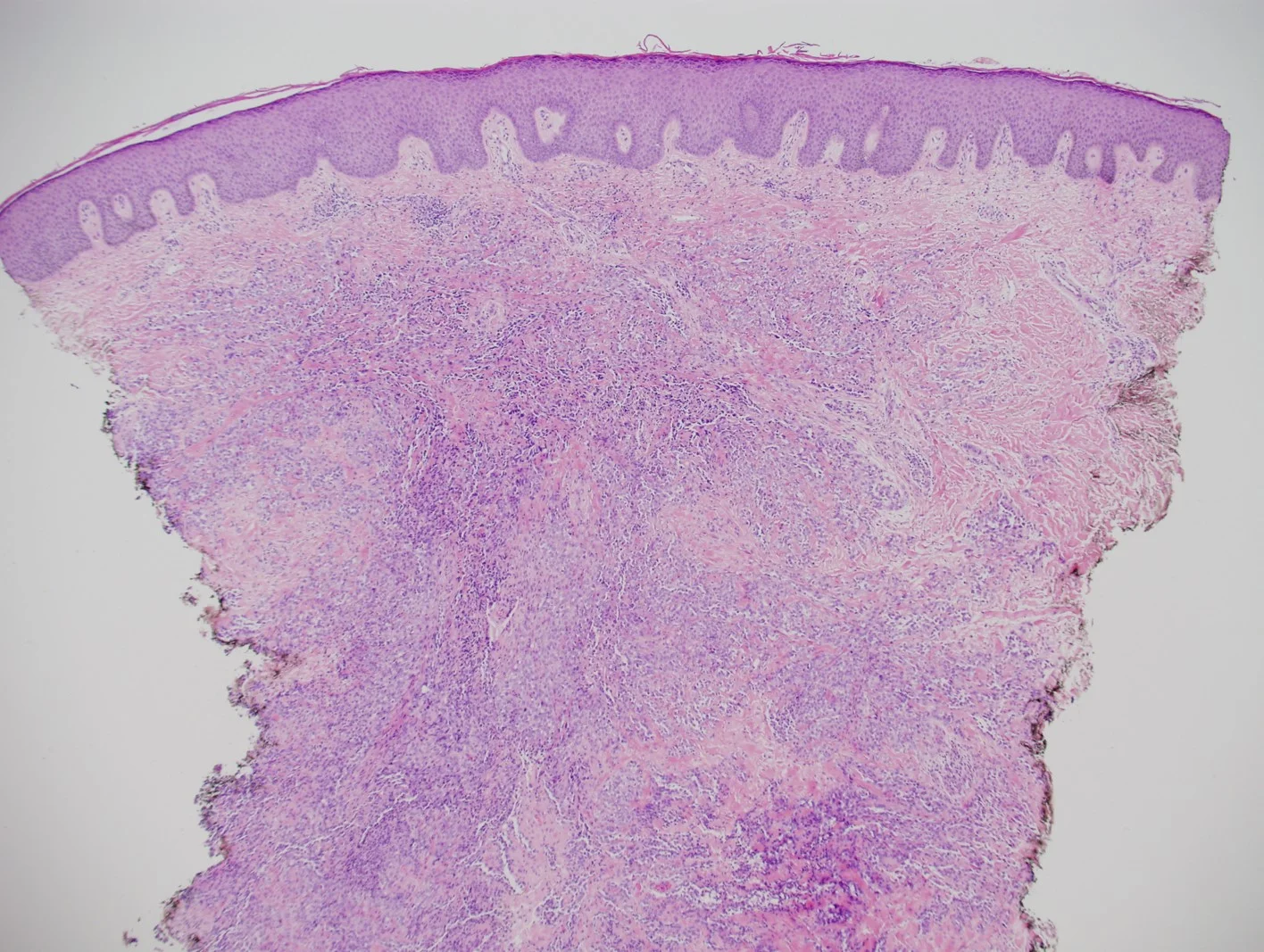



Case 15B: Unusual Pigmented Epithelioid Angiosarcoma Simulating Melanoma
Caregivers: Adean Kingston, MD, Etan Marks, DO, Clay J. Cockerell, MD; Dallas, TX
History: A 73 year old male presented with a long standing lesion on the occipital scalp. A shave biopsy was submitted as “concerning for melanoma.”
Physical Examination: Irregular dark pigmented lesion on the scalp within an area of diffuse erythema extending onto the forehead.
Histopathology: There was a dense diffuse infiltrate of spindle and epithelioid cells with pigment scattered throughout the lesion. Most of the neoplastic cells were closely apposed to one another and some were arranged in aggregations. There were a few were few dilated blood vessels but no irregular staghorn vessels with atypical cells in their lumina were noted. A diagnosis of malignant melanoma >2.1 mm was initially rendered.
The lesion was re-excised at which point features characteristic of angiosarcoma were identified with numerous, diffuse atypical irregular vessels with endothelial atypia seen throughout the dermis.
The submitting clinician was contacted and a request was made to review clinical photographs. Immunohistochemical stains were performed on the original biopsy and stains for ERG and CD31 were positive while stains for Melan-A and SOX10 were negative. Of interest, one popular area originally sampled by shave technique stained positively for S-100 protein.
Diagnosis: Pigmented Epithelioid Angiosarcoma Simulating Melanoma
Clinical Course: A re-excision was performed although there was diffuse involvement of lateral margins given the diffuse nature of the process. He was referred to radiation oncology for post-operative palliative radiotherapy.
Points of Emphasis: Angiosarcoma is an uncommon soft tissue neoplasm with a predilection for skin and superficial soft tissues. The epithelioid variant of angiosarcoma can sometimes be difficult to recognize and can mimic other neoplasms, usually epithelial lesions such as squamous cell carcinoma. In this case because the lesion did not demonstrate significant vascular differentiation and had pigment which was probably hemosiderin, in the context of a clinical history of possible melanoma, the diagnosis of melanoma was made on the basis of morphology alone. It was only apparent on re-excision that the lesion truly represented angiosarcoma.
Of interest in this case, there was positive staining for S-100 protein which may be seen in some some angiosarcomas which can further complicate the diagnosis. Furthermore, the clinician noted only the pigmented papule and did not notice the larger, diffuse erythema which represented the more classic angiosarcoma.
It is important for clinicians to consider angiosarcoma in the differential diagnosis of cutaneous malignancies in lesions on the scalp of older individuals, especially if there are erythematous, macular “bruise-like” areas. Shave biopsies are often subject to sampling errors and malignancies such as angiosarcoma and other soft tissue neoplasms may demonstrate multiphasic patterns that may appear quite different within the same neoplasm. In this case, the biopsy sampled an epithelioid component that was not characteristic of angiosarcoma although it was recognizable as a malignancy. Unfortunately, the treatment of angiosarcoma is difficult as the lesion is diffuse and is not usually surgically resectable. Radiation therapy including brachytherapy may be useful for palliation although the prognosis remains extremely poor.
References: 1. Prescott RJ, Banerjee SS, Eyden BP, Haboubi NY. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology 1994;25 (5): 421–429. 2. Bacchi CE, Silva TR, Zambrano E, et al. Epithelioid angiosarcoma of the skin. Am J Surg Pathol 2010;34:1334–43















Case 16: Glomangiosarcoma
Caregivers: Marisa Belaidi, M.D., Trent Massengale, M.D, New Orleans, LA
History: An 83 y.o man with a history of non-melanoma skin cancer presented to clinic for a “spot” on his scalp that was present, reportedly for weeks.
Physical Examination: Anterior scalp: erythematous scaly plaque
Histopathology: Diffuse proliferation of cells with round oval nuclei and abundant cytoplasm. Most of the cells are present around vascular structures. Scattered mitotic figures are noted. Immunoperoxidase stains for vimentin are positive throughout this proliferation.
Clinical Course: The patient was sent for Mohs surgery, during which time the lesion was completely excised and margins cleared. A skin graft was placed following clearance of the tumor. The patient has returned for follow-up without recurrence.
Diagnosis: Glomangiosarcoma
Points of Emphasis: First described in the literature by Lumley and Stansfeld in 1972, malignant glomus tumors, also known as glomangisarcomas, are exceedingly rare vascular neoplasms, most commonly located on the distal extremities1-2. Few have been reported to exhibit metastatic potential, though it has been documented2. Diagnosis is confirmed via histology, which may show a variable degree of nuclear atypia, necrosis and/or mitotic activity. Other potential worrisome signs for a tumor comprised of glomus cells includes tumor with a deep location and size > 2 cm2. A common feature of both benign and malignant glomus cell tumors is distinct cell borders. Immunohistochemistry is useful in ruling out other neoplasms in the differential. Glomangiosarcomas will stain positive for vimentin and smooth muscle actin, and are negative for other markers including S100 and epithelial markers3. Our case exhibited histologic findings described in malignant glomus tumors and IHC provided confirmation.
Mainstay of therapy for glomangiosarcoma is complete surgical excision3.
References: 1. Woodward JF, Jones NF. Malignant Glomus Tumors of the Hand. Hand 2016;11(3):287-289. 2. Folpe AL, Fanburg-Smith JC, Miettinen M, Weiss SW. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25(1):1-12 3. Rishi A, Dulanto F, Chen S. Glomangiosarcoma in the shoulder of a 51-year-old man. Dermatol Pract Conc. 2012;2(1):9.







Case 17: Cutaneous Endometriosis of the Umbilicus(Villar’s Nodule)
Caregivers: Margaret Brown, MD, Tracy Biediger, MD; San Antonio, TX
History: A 26 year-old Caucasian nulliparous female with no pertinent dermatologic history presented with a tender growth in her umbilicus that had been present for 2 years. She experienced monthly bleeding from the lesion. She has never had abdominal surgery but did have her umbilicus pierced in the remote past though no longer wears jewelry. Of note, she also reported debilitating dysmenorrhea and menorrhagia. Her last menstrual period was 2 weeks prior to our exam and biopsy.
Physical Examination: Physical exam reveals a 4 mm brown to violaceous delicate bi-lobed papule within the umbilicus. It was sensitive to light touch and palpation.
Laboratory Data: Complete blood count (CBC) and iron studies were within normal limits.
Histopathology: A snip biopsy reveals glandular spaces within a fibrovascular stroma. Some of the glandular cells are exhibiting decapitation secretion. The biopsy is consistent with endometriosis. CD10 and ER IHC stains support the diagnosis.
Clinical Course: The patient was referred to gynecology for evaluation of endometriosis. She is pending initial consultation with gynecology and will follow-up with Dermatology.
Diagnosis: Cutaneous Umbilical Endometriosis (Villar’s Nodule)
Points of Emphasis: Cutaneous endometriosis (CEM) arising in the umbilicus is also known as Villar’s nodule. Endometriosis is common and has been reported in virtually every organ system. Several hypotheses have been proposed regarding its pathogenic mechanism including metaplasia, venous metastasis, lymphatic metastasis, and iatrogenic implantation.1 CEM is rare, accounting for 0.1-5.5% of all endometriosis. It typically occurs in the setting of open surgical or laparoscopic procedures but less often occurs spontaneously. When occurring spontaneously, CEM shows a predilection for the umbilical or inguinal regions.2 In our patient, her CEM was in the setting of remote umbilical piercing, but did not appear to involve the scar itself.
Clinically, CEM presents as a tender brown, red, or blue papule or nodule. Its appearance may fluctuate with the hormonal environment, and it may demonstrate catamenial bleeding. The differential diagnosis of Villar’s nodule includes nevus, melanoma, Sister Mary Joseph nodule, cutaneous metastasis, keloid, urachal duct cyst, cutaneous endosalpingiosis, and pyogenic granuloma.2 While the clinical presentation may be sufficient to make the diagnosis, a biopsy is required to rule out these other potentially serious entities.
Common histopathologic features of CEM include dilated endometrial glands lined by pseudostratified columnar epithelium in a fibrovascular stroma. Cellular nuclei of glandular cavity walls may be positive on immunohistochemical staining for estrogen and progesterone receptors, and cells in the interstitium may be positive for CD10 (sensitive for endometrial stroma).3
Definitive treatment of cutaneous endometriosis includes wide surgical excision; a 2 mm margin is reasonable. Hormonal therapy can also be considered prior to surgical excision to decrease bleeding.1 Malignant transformation is very rare but has been reported.4 Patients should be evaluated by Gynecology.
References: 1. Din AH, Verjee LS, Griffiths MA. Cutaneous endometriosis: A plastic surgery perspective. Journal of Plastic, Reconstructive and Aesthetic Surgery 2013;66 (1), 129-130. 2. Kyamidis K, Lora V, Kanitakis J. Spontaneous cutaneous umbilical endometriosis: report of a new case with immunohistochemical study and literature review. Dermatol Online J. 2011;17(7):5. 3. Fukuda H, Mukai H. Cutaneous endometriosis in the umbilical region: The usefulness of CD10 in identifying the interstitium of ectopic endometriosis. Journal of Dermatology 2010; 37:545–549. 4. Shalin SC, Haws AL, Carter DG, Zarrin-Khameh N. Clear cell adenocarcinoma arising from endometriosis in abdominal wall cesarean section scar: A case report and review of the literature. J Cutan Pathol 2012;39:1035-41.




Case 18: Tattoo Reaction with Pseudo-Epitheliomatous Hyperplasia to Red Pigment
Caregivers: Robert Christopher Gilson, MD, Sarah Groff, MD, Sandra Osswald, MD; San Antonio, TX
History: 46-year-old female with no significant past medical history presenting for evaluation of rash to her right foot. She states she had this tattoo placed 1-2 months prior to it becoming red and tender. After getting a vine tattoo on her right foot, it was initially swollen. The swelling went down for about 1 month. 2 months later, it became swollen, painful and hot to the touch. The erythema and pain were confined to the red area of the tattoo only. She was prescribed cephalexin for 2 weeks, which did not help. She has also tried bacitracin, triple antibiotic ointment, and most recently was using a “soothing itch” cream from Mexico.
Physical Examination: Indurated erythematous/violaceous nodules with overlaying scale and surrounding erythema only within only the red areas of her tattoo on R dorsal foot
Laboratory Data: Tissue submitted to micro for Gram, AFB, and fungal stains were all negative; fungal and mycobacterial tissue cultures were negative at 6-week final.
Histopathology: Pseudoepitheliomatous hyperplasia with chronic inflammation and tattoo ink; GMS, fungal PAS, AFB, and Gram stains negative for organisms.
Clinical Course: There is some surrounding erythema to each of the areas that had red ink coloring, and given the pain and inflammation, an infection was considered in addition to a granulomatous tattoo reaction to the red ink. The patient was prescribed clarithromycin 500mg twice a day to cover several of the previously reported tattoo related atypical mycobacteria infection which would cover for both Mycobacterium chelonae and M. abscessus, both of which have been reported with tattoo site infections and can be found in the tap water often used to dilute tattoo inks. The patient never took the clarithromycin as she felt antibiotics had not been helpful. She was seen in follow up and subsequently treated with IL TAC 10mg/cc after cultures were negative.
Diagnosis: Tattoo reaction with pseudo-epitheliomatous hyperplasia to red pigment
Points of Emphasis: Pseudo-epitheliomatous hyperplasia (PEH) pattern can mimic squamous cell carcinoma as well as possible infection with atypical mycobacteria. There are numerous reports of atypical mycobacteria in the literature due to contaminated dyes that have been mixed with tap water.
Red pigment is the ink most commonly associated with adverse reactions. Previously this was due to the use of cinnabar (mercuric sulfide) but now its use has been replaced by cadmium selenide, sienna, ferric hydrate (red ochre) and organic dyes1. Mycobacterial infections can occur as early as 1-month post tattoo and if a granulomatous lesion is noted, biopsy is warranted for tissue culture and possible PCR for mycobacterial typing. Early antibiotic therapy is recommended rather than awaiting culture results. Once an infection is confirmed, therapy often needs to be continued for several months. Antibiotics can be adjusted based on culture results.
PEH is a relatively rare histologic finding in tattoo reactions. Non-granulomatous cutaneous inflammatory tattoo reactions present as a number of different patterns including spongiotic, psoriasiform, interface, vesiculobullous, vasculitis, fibrosing and pseudoepitheliomatous. Other histologic patterns include nodular and diffuse granulomatous infiltrates that may be tuberculoid, sarcoidal, suppurative, necrobiotic as well as pseudolymphomatous2.
There are few reported cases of PEH confined to red tattoo areas. It typically presents clinically as rapidly growing verrucous growths occurring 1 week to several months after the tattoo placement2. There is a spectrum of epidermal reactions from benign pseudoepitheliomatous hyperplasia to keratoacanthoma and rare squamous cell carcinoma. Distinguishing PEH tattoo reaction from a genuine SCC can be difficult. PEH typically presents with bland cytologic features and lack nuclear crowding, pleomorphism and atypical mitoses. It is important to note any history of tattoo placement to prompt the possibility of a non-neoplastic etiology such as tattoo dye reaction to avoid over-diagnosis as a neoplasm. Noting tattoo pigment in tissue section is also helpful in making an accurate diagnosis. As noted in a 2016 review article, 50 cases of skin cancer over 40 years, have been reported arising in tattoos (23 SCC and KA, 16 Melanoma, 11 BCC). An overall estimated incidence of cancer in a tattoo is felt to be under 1 in 200 million2.
Treatment for tattoo reactions can include topical or intralesional steroids, laser, and surgical removal.
References: 1. Islam, Parvez S. Medical Complications of Tattoos: A Comprehensive Review. Clin Rev Allergy Immunol. 2016 Apr;50(2):273-86. 2. Thum CK, Biswas, A. Inflammatory complications related to tattooing: a histopathological approach based on pattern analysis. Am J Dermatopathol. 2015 Jan;37(1):54-66.





Case 19: Nodular Amyloidosis
Presenters: Havens Cary, MS IV, Jason L. Smith, MD; Rome, GA; New Orleans, LA
History: 55-year-old male presents with a one year history of a solitary lesion on the left corner of his mouth. He denies similar lesions elsewhere.
Medical History: The patient’s past medical history is significant for extensive exposure to solar radiation. Patient endorses occasional alcohol and daily tobacco use.
Physical Examination: 1.1 cm erythematous, tumor nodule involving the left oral commissure.
Laboratory Data: CBC: WNL, CMP: WNL, SPE: WNL
Histopathology: Dense dermal deposits of glassy eosinophilic material replace adnexal structures with extension into underlying subcutaneous fat. Mild to moderate degree of reactive plasmacellular inflammation surrounds the periphery of the eosinophilic material, mainly concentrated within the papillary dermis. The overlying epidermis is characterized by a moderate degree of reactive hyperplasia and hyperkeratosis.
Congo Red stain displays areas of apple green birefringence under polarization consistent with amyloid. An immunohistochemical stain for CD138 in combination with in situ hybridization stains for both kappa and lambda confirm the presence of a polyclonal population of plasma cells.
Clinical Course: The lesion has not changed significantly with no associated itching or sloughing of tissue.
Diagnosis: Nodular amyloidosis
Points of Emphasis: Nodular amyloidosis is the rarest of the 3 forms of primary cutaneous amyloidosis. Unlike macular and lichen amyloidosis, nodular amyloidosis is derived from immunoglobulin light chains. Our patient presented with solitary nodular amyloidosis in the left corner of mouth that has been persistent for one year.
References: 1. Moon AO, Calamia KT, Walsh JS. Nodular AmyloidosisReview and Long-term Follow-up of 16 Cases. Arch Dermatol. 2003;139(9):1157–1159. doi:10.1001/archderm.139.9.1157 2. Cornejo, Kristine M., Frances J. Lagana, and April Deng. "Nodular amyloidosis derived from keratinocytes: an unusual type of primary localized cutaneous nodular amyloidosis." The American Journal of Dermatopathology 37.11 (2015): e129-e133.

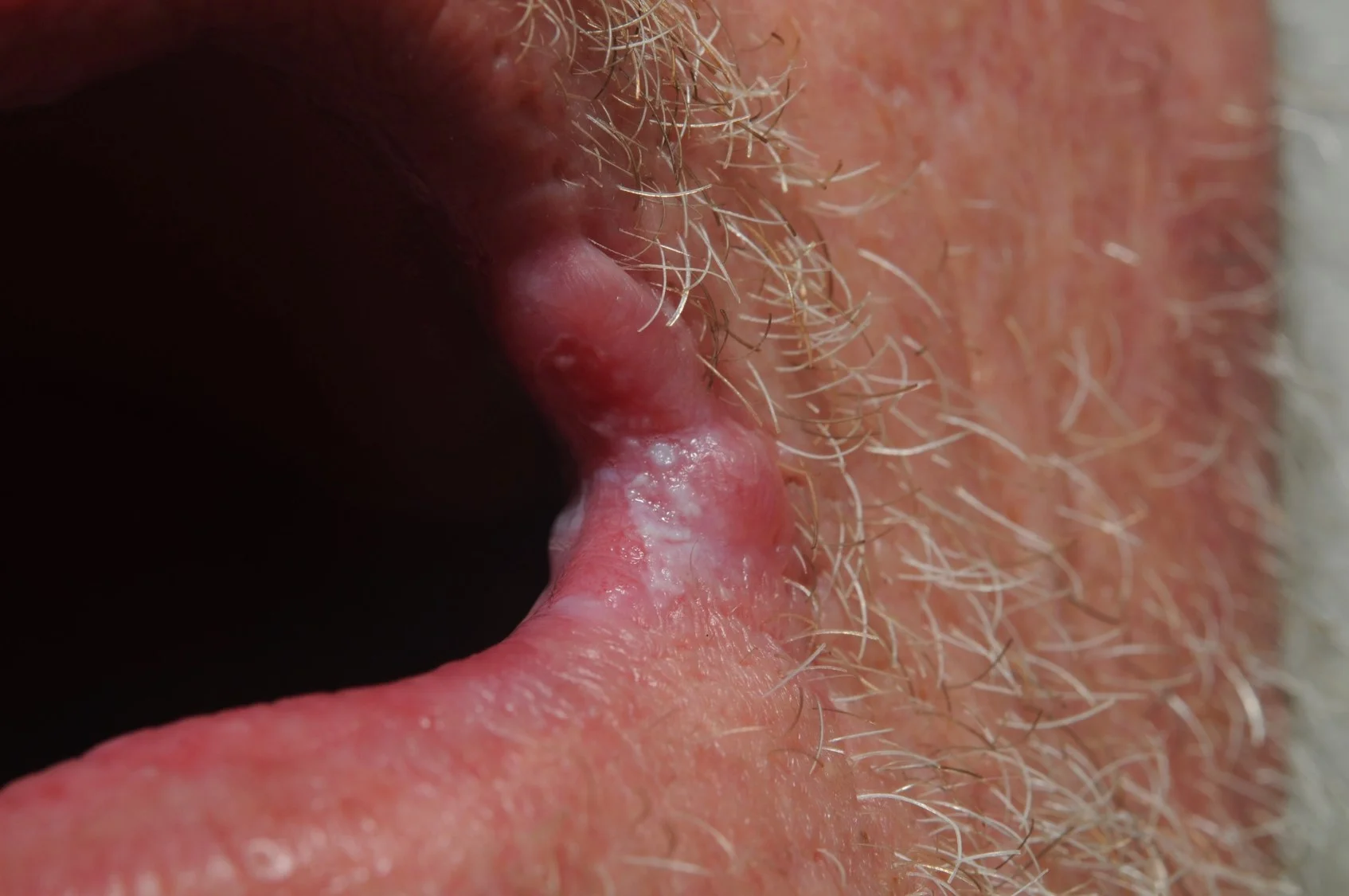




Case 20: Migrating Filler
Caregivers: Caroline Lee, MD, Pamela C. Martin, MD, Sharon S. Meyer, MD, Hope G. Martin MS-I; New Orleans, LA
History: A 73 year-old female presented with the chief complaint of a new bump on her lower face. She was last seen 10 months ago for injection of calcium hydroxyapatite into her cheeks as well as hyaluronic acid (Volbella®) injected into her upper and lower mucosal lip. She was previously injected with other hyaluronic acid fillers 6 years ago. This was her first and only treatment with this particular hyaluronic acid filler. She adamantly denies ever receiving filler in the location of the new lesion.
Physical Exam: Left lower cutaneous lip with a 1 cm firm, movable, non-tender subcutaneous nodule with overlying erythema, no visible puncta. Left upper cutaneous lip with crusted pink papule
Histopathology: Amorphous basophilic, non-polarizable material is present in the dermis and subcutis both freely and in association with palisaded histiocytes compatible with hyaluronic acid.
Clinical Course: After biopsy confirmed the presence of hyaluronic acid filler, the patient was started on doxycycline 100mg daily for 3 weeks and the lesions were injected with hyaluronidase on two separate occasions. At 2 month follow-up the lesions were much improved.
Diagnosis: Migrating filler
Points of Emphasis: Filler with hyaluronic acid is one of the most commonly performed nonsurgical procedures for facial soft tissue augmentation due to its biocompatibility, biodegradability, and rapid recovery time. While soft tissue fillers are considered relatively safe and non-invasive, they are foreign bodies that may result in a variety of complications. Most adverse reactions occur shortly after injection but others may occur months to years after injection. Notable complications include nodules from uneven distribution of the filler, inflammation, itching, bruising, bluish discoloration (Tyndall effect), necrosis, hypersensitivity reactions, foreign body granuloma and migration.
Filler migration represents the presence of filler at a location remote from the primary injection site and can occur with many different types of filler other than hyaluronic acid. There are several mechanisms by which filler can travel to other sites which include injection technique- related, vigorous massage after injection, muscle activity or dislocation of filler, gravity, pressure-induced displacement or dislodgement, lymphatic spread, and intravascular injection. Of note, injection technique –related migration encompasses poor technique of the injector resulting in filler inadvertently placed in an adjacent site to the intended one, high volume of filler resulting in overflow or filler injected under pressure resulting in movement to a nearby area. Furthermore, it is important to remember that each of the described mechanisms of migration does not occur with all types of filler.
Although use of fillers is generally safe with minimal side effects, physicians should be aware of filler migration to sites remote from injection and the persistence of filler with presentation as new nodules even years after initial injection. It is important that physicians inquire about past soft tissue fillers during consultation, ideally with filler name and injection site. Hyaluronidase can be used to treat misplaced filler.
References: 1. Jordan D. and Stoica B. Filler Migration: A Number of Mechanisms to Consider. Ophthalmic Plastic and Reconstructive Surgery. 2015; 31(4):257-262 2. Lee M, Sung M, Kim N, Choung H, and Khwarg S. Eyelid Mass Secondary to Injection of Calcium Hydroxyapatite Facial Filler. Ophthalmic Plastic & Reconstructive Surgery. 2008; 24(5): 421-423. 3. Chae, S., Lee, K., Jang, Y., Lee, S., Kim, D. and Lee, W. (2016). A Case of the Migration of Hyaluronic Acid Filler from Nose to Forehead Occurring as Two Sequential Soft Lumps. Annals of Dermatology. 2016; 28(5): 645. 4. Lin, C., Chiang, C., Wu, B. and Gao, H. Filler Migration to the Forehead due to Multiple Filler Injections in a Patient Addicted to Cosmetic Fillers. Journal of Cosmetic and Laser Therapy. 2017; 19(2):124-126. 5. Jones, D. Update on Emergency and Nonemergency Use of Hyaluronidase in Aesthetic Dermatology. JAMA Dermatology. 2018; 154 (7): 763-764.


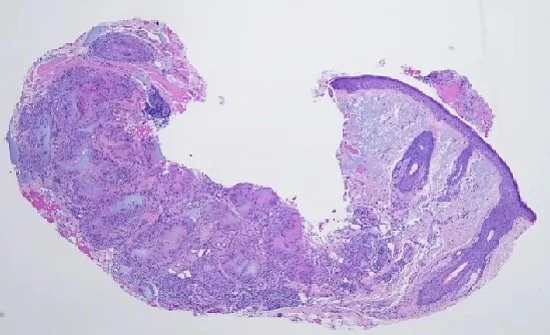




Case 21a: Eosinophilic Fasciitis
Caregivers: John P. Miller, MS IV, Jason L. Smith, MD; New Orleans, LA; Rome, GA
History: A 54-year-old white female presents with swelling and sensation of tightness of the skin of the bilateral upper extremities for 3 months. She stated that the swelling/tightness had recently spread to her trunk and proximal lower extremities. She also experienced a 30 pound unintentional weight gain over this same period. She noted minimal improvement with Lasix 20mg qd. She denied pruritis or pain of lesions and had no fever, fatigue or night sweats. She has experienced increased shortness of breath due to what she describe as tightness of her chest and abdomen.
Medical History: COPD (3ppd), HLD, osteopenia
Physical Examination: Skin with mild erythema and sclerotic changes most prominent of the skin of the inner arm and forearm. There was woody induration appreciated with no tenderness to palpation. There was diffuse skin involvement affecting the neck, trunk, and bilateral upper extremities with sparing of face, hands, feet, BLE, and genitalia.
Laboratory Data: Eosinophilia 1.7H (0-0.4), otherwise normal CBC/CMP and ESR. ANA and SCL-70 negative. SPEP shows faint band in gamma region with slight M-spike 0.3 g/dL.DEXA -2.7 at L4, Abnormal pulmonary function studies were found as well.
Histopathology: Excisional biopsy of right inner arm revealed deep dermal and subcutaneous thickening with prominent fibrous septa. Within the septa there are increased numbers of inflammatory cells including neutrophils, eosinophils, and histiocytes. There were prominent eosinophils. Eosinophils also involved the deep dermis. The histology is most consistent with eosinophilic fasciitis.
Clinical Course: Patient achieved significant improvement initially while on oral prednisone taper 60mg but due to worsening osteopenia patient, this was discontinued and she was started on Methotrexate 15 mg weekly. She experienced complete resolution in one year.
Diagnosis: Eosinophilic Fasciitis
Points of Emphasis: Eosinophilic fasciitis is characterized in its early phase by erythema and edema of the limbs or trunk and later by collagenous thickening of the subcutaneous fascia. Eosinophilia is often a prominent laboratory finding in the early phase and may become less prominent in later stages of disease. Systemic sclerosis, Churg-Strauss disease, peripheral T-cell lymphoma, and malignancy among others should be ruled out via clinical and immunologic studies. Systemic corticosteroids are efficacious and the current standard of therapy.
References: 1. Lebeaux, D. & Sène, D. Eosinophilic Fasciitis (Shulman Disease). Best Practice & Research Clinical Rheumatology. August 2012; 449-458. 2. Pinal-Fernandez, I.; Selva-O’Callaghan, A.; Grau, J.M. Autoimmunity Reviews. January 2014; 379-382.









Case 21B: Bullous Morphea
Caregivers: John L. Millns, MD, Laura Millns, PA-C, Charles Perniciaro, MD; Tampa, FL
History: This 61-year-old fair-complexioned female presented with a chief complaint of thickening of the skin and focal blistering of the right and left thigh, chest, periaxillary region, abdomen, left breast, and upper back of approximately 7 months duration. The patient claims that she was under a tremendous amount of psychological stress about the time the skin lesions began.
Physical Examination: On examination, the patient was found to have bound-down hyperpigmented plaques involving the anterior proximal thighs, abdomen, anterior chest, periaxillary area, and upper back. Blistering areas were identified on the thigh and hip areas. More significant sclerosis and hyperpigmentation were identified on the upper back adjacent to areas of the sports bra, while sparing was noted on the areas protected by the sports bra.
Laboratory Data: CBC with differential normal. Metabolic panel normal except ALT (SGPT) elevated at 40 IU/L. Urinalysis normal. Lipid panel: Cholesterol elevated at 293 mg/dL, LDL elevated at 154 mg/dL. Thyroid panel normal. Lyme titer: IgG and IgM by Western Blot negative, ANA negative, anti-SS-A and anti-SS-b negative, antiscleroderma-70 antibodies negative. Hepatitis C virus antibody negative.
Histopathology: 6/18/18: Right anterior proximal thigh – marked papillary dermal edema with dilated lymphatics and deep dermal sclerosis (consistent with morphea), with a predominantly lymphocytic inflammatory reaction and occasional eosinophils. 7/10/18: Right superior upper back, involved skin – superficial and deep dermal sclerosis with a predominantly lymphocytic perivascular and interstitial inflammatory reaction with plasma cells, characteristic of morphea. 7/10/18: Right superior upper back, clinically normal skin – superficial and deep dermal sclerosis with perivascular and interstitial lymphocytic inflammatory reaction with an admixture of plasma cells, characteristic of morphea. 8/17/18: Punch biopsies from the right abdomen and right anterior proximal thigh representing areas of normal skin with faint erythema but no induration, showing histologic features of morphea with associated subacute spongiotic dermatitis with eosinophils consistent with secondary atopic dermatitis or allergic contact dermatitis.
Clinical Course: The patient was treated with several courses of prednisone taper from 40 mg for over 3 weeks with no effect on the morphea. After only 2 weekly doses of methotrexate 15 mg per week, the patient reported softening of her skin.
Diagnosis: Bullous morphea.
Points of Emphasis: Bullous morphea may coexist with non-bullous areas of morphea. Areas of normal-appearing skin may show significant histologic changes of morphea. Sun exposure may exacerbate the changes of morphea.
References: 1. Daoud MS, Su WP, Leiferman KM, Perniciaro C. Bullous morphea: Clinical, pathological and immunopathologic evaluation of thirteen cases. J Am Acad Dermatol 1994 July;30:937-43. 2. Trattner A, David M, Sandbank M. Bullous morphea: A distinct entity? Am J Dermatopathol 1994 August; 16(4):414-7. 3. Angel Fernandez-Flores, et al. Three cases of bullous morphea: Histopathologic findings with implications regarding pathogenesis. Journal of Cutaneous Pathology, volume 42, #2, February 2015, pages 144-149.































Case 22: Morbihan’s Disease
Caregivers: Drew Kuraitis, Aimee Coscarart, Alun Wang, MD, Laura Williams; New Orleans, LA
History: A 32-year old woman presened with persistent upper facial redness and swelling for the past 9 years. She was biopsied on the cheek and diagnosed with rosacea after her rash first presented. She tried various topical agents and oral antibiotics without improvement. She was again biopsied in 2017, this time samples were taken from each of her eyelids. Pathology from one lid biopsy was consistent with Melkersson-Rosenthal Syndrome (MRS). She was then treated with a 3 to 4 month course of high dose ibuprofen, dapsone, hydroxychloroquine and a trial of methotrexate, none of which provided any improvement in symptoms. She denies swelling or edema of her lower face or lips, and also denies any history suggestive of facial neuropathy.
Physical Examination: Pleasant woman in no acute distress. Skin exam is notable for well-demarcated erythematous plaques on the forehead and bilateral cheeks with scattered erythematous papules and pustules. All of her eyelids are erythematous, soft, edematous and non-tender, with the right upper lid most affected. Mild fissuring of tongue. Skin scraping is positive for Demodex.
Laboratory Data: CBC, CMP, TSH, A1C, lipid panel within normal limits.
Histopathology: 10/2017: Right upper eyelid. Dermal stromal edema with lymphangiectasia, perivascular lymphocytes and intralymphatic histiocytosis. The histologic findings are compatible with Melkersson-Rosenthal syndrome. 9/2018: Forehead. Perivascular and perilymph granulomatous accompanied by prominent telangiectasis. There is prominent edema of dermis with lymphangiectasia and intralymphatic histiocytosis. A perivascular and perifollicular infiltrate of lymphocytes and plasma cells is present. Demodex mites are present in the hair follicles. Sebaceous hyperplasia is present. There are focal calcium deposits in the dermis, consistent with calcinosis cutis. Mixed features of granulomatous rosacea and Melkersson-Rosenthal syndrome are appreciated.
Clinical Course: Erythema improved after 2 doses of oral ivermectin and spironolactone 50mg to 100mg daily. She has been consented to start isotretinoin.
Diagnosis: Morbihan’s disease.
Points of Emphasis: Morbihan’s disease (MD) is a rare complication of patients with rosacea, characterized by persistent facial edema of the eyelids, forehead, glabella and cheeks[1]. Histologic findings are often non-specific, including dermal edema, dilated vessels, perivascular and perifollicular infiltrates consisting of histiocytes, neutrophils and lymphocytes. One report identified perilymphatic granulomas with intralymphatic histiocytosis, suggesting physical disruption of dermal lymphatics, leading to lymphedema[2]. A recent case series of 5 patients diagnosed with MD was able to re-demonstrate classic findings of rosacea, as well as perilymphatic infiltrates in the dermis in addition to dilated lymphatic channels, evidenced by D2-40 staining[3]. Our patient’s recent biopsy from the forehead demonstrated similar findings, including perivascular and perilymphatic granulomatous inflammation, and intralymphatic histiocytosis. MRS is often diagnosed as a triad of facial (typically orofacial) swelling, facial paralysis and a fissured tongue. Pathology is often non-specific, showing non-caseating perilymphatic and perivascular granulomas with lymphatic dilation[4]. Although the pathologic findings are similar between MD and MRS, our patient’s lack of neuropathic symptoms, location on the upper face, and her history of rosacea are suggestive of the the entity of MD. Both conditions are often refractory to treatment with steroids and immunomodulating agents, but there are reports of MD being successfully treated with isotretinoin, which is what our patient is planning to start.
References: 1. Jansen T. and Plewig G. The treatment of rosaceous lyphoedema (1997). Clin Exp Dermatol 22:57. 2. Nagasaka T, et al. Persistent lymphoedema in Morbihan disease: formation of perilymphatic epithelioid cell granulomas as a possible pathogenesis. (2008). Clin Exp Dermatol. 33:764-67. 3. Carruth, et al. Extreme eyelid lymphedema associated with rosacea (Morbihan disease): Case series, literature review, and therapeutic considerations (2017). Ophthal Plast Reconstruct Surg 33(35):S34-6. 4. Cockerham et al. Melkersson-Rosenthal syndrome: new clinicopathologic findings in 4 cases (2000). Arch Ophthalmol 118:227-32.





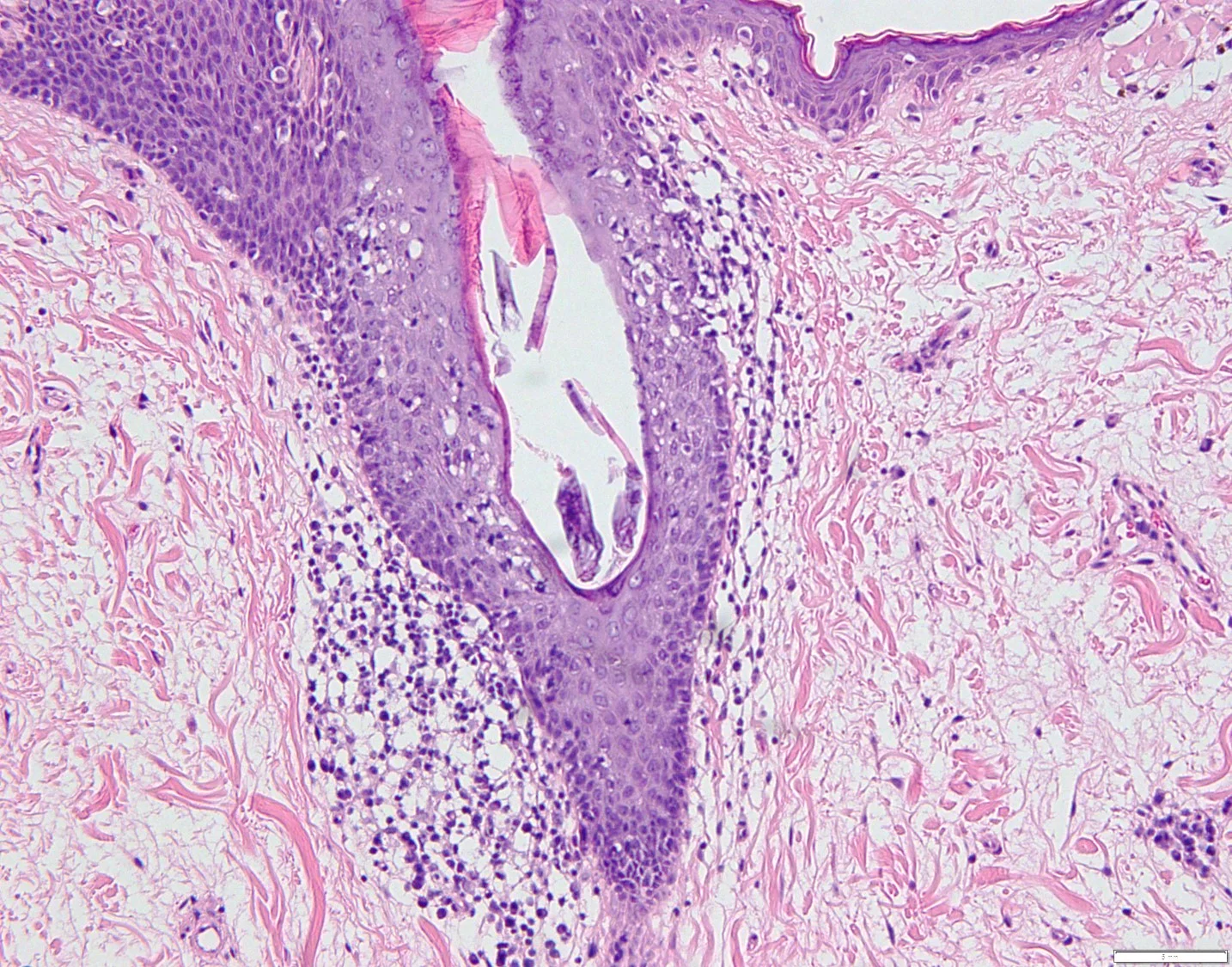


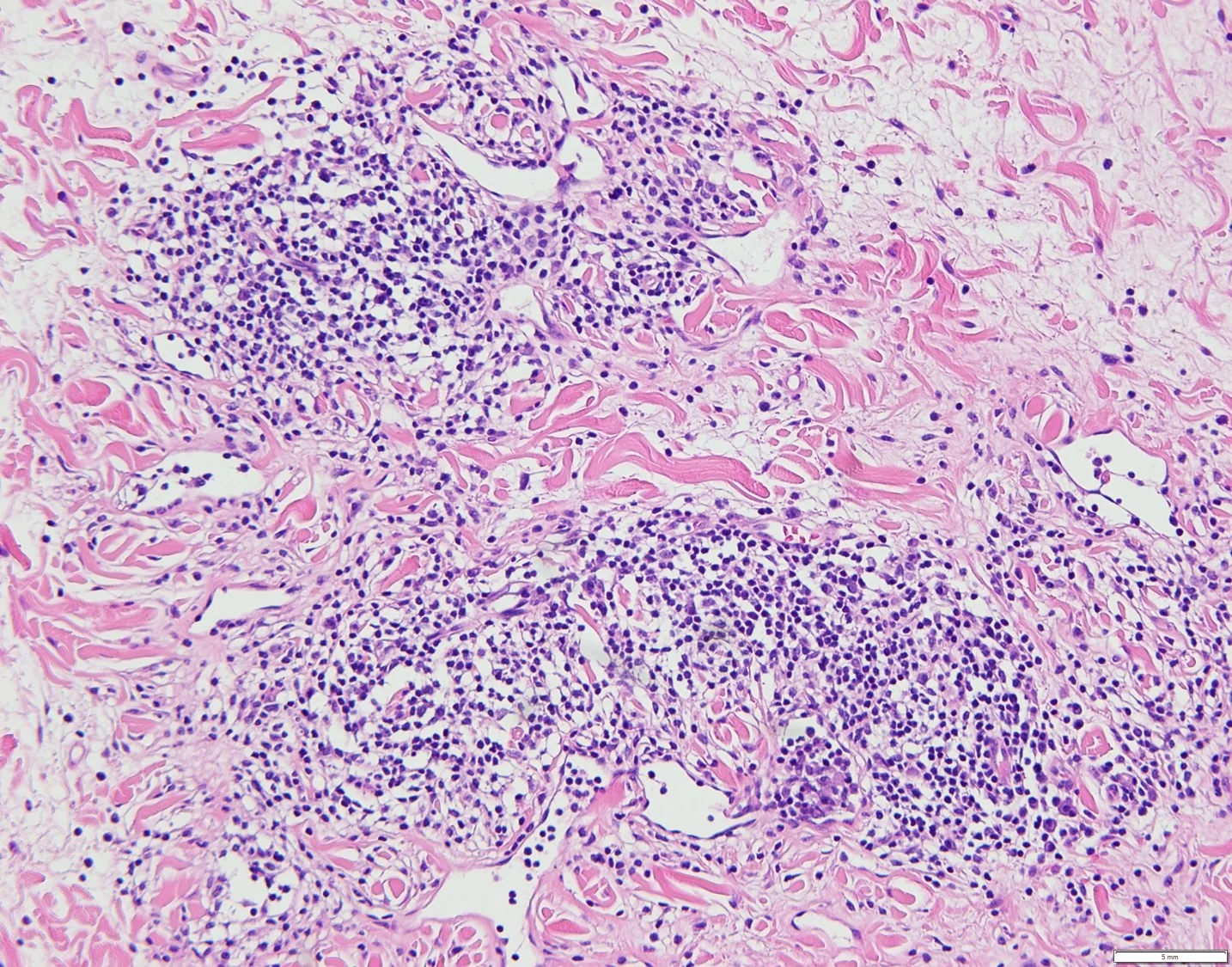

Case 23: Pityriasis Rubra Pilaris with Acantholysis
Caregivers: Travis Vandergriff, MD; Ilka Netravali, MD; Heather Goff, MD; Dallas, TX
History: A 58 year old Caucasian man presented to the dermatology clinic for evaluation of a rash which had been present for about one year. He first noted redness and swelling in his legs, along with a scaly and pruritic eruption on the upper body. The rash generalized and involved most of the body. He had no history of asthma or atopy as a child. He was diagnosed with atopic dermatitis by a referring physician and was treated with dupilumab with no improvement.
Physical Examination: Nearly confluent, erythematous thin scaly plaques on the arms and legs, with erythematous scaly papules (some follicular) on the back and abdomen.
Histopathology: Two biopsies were performed, both of which showed psoriasiform epidermal hyperplasia irregular acanthosis and hyperkeratosis with small foci of spongiosis. Both biopsies also demonstrated foci of acantholysis, with some dyskeratosis as well.
Clinical Course: Patient was treated with triamcinolone ointment wet wraps and emollients, with some improvement. He will soon begin treatment with acitretin.
Diagnosis: Pityriasis rubra pilaris with acantholysis
Points of Emphasis: Pityriasis rubra pilaris is an uncommon papulosquamous eruption with several different clinical variants having been described. The most common type (Type I) presents in adults with follicular-based, hyperkeratotic papules that spread inferiorly after starting on the upper body. Patients often develop palmoplantar keratoderma. An atypical presentation in the adult involves follicular hyperkeratotic papules, with an eczematous or ichthyosiform appearance to the plaques on the legs. These patients often have a more generalized distribution with chronic disease. The histological findings in pityriasis rubra pilaris vary by the acuity of the eruption and are most characteristic during the acute phase. Classic features include irregular acanthosis and a cornified layer with alternating orthokeratosis and parakeratosis. Follicular ostia are plugged with keratin, and inflammation is sparse. Foci of acantholysis, or acantholytic dyskeratosis, may be seen in approximately 20% of cases of pityriasis rubra pilaris.
In this case, the original clinical history did not include a differential diagnosis of psoriasis or PRP and the main histologic features identified were the acantholytic dyskeratosis which led to an initial diagnosis of Grover’s disease. When the patient was presented at a clinical conference, it became apparent that the psoriasiform dermatitis was the most important feature and a diagnosis of PRP with acantholysis was made. This emphasizes the importance of providing complete clinical information when submitting skin biopsies and in difficult cases, including clinical photographs or having the patient evaluated at a clinical conference as was done in this case can improve the accuracy of the diagnosis.
References: 1. Avitan-Hersh E, Bergman R. The Incidence of Acantholysis in Pityriasis Rubra Pilaris-Histopathological Study Using Multiple-Step Sections and Clinicopathologic Correlations. Am J Dermatopathol. 2015 Oct;37:755-8. 2. Magro CM, Crowson AN. The clinical and histomorphological features of pityriasis rubra pilaris. A comparative analysis with psoriasis. J Cutan Pathol 1997;24:416-24.
















Case 24: Psoriasiform Cutaneous Eruption Associated with Pembrolizumab Therapy
Caregivers: Leah Persad, DO, Kevin Honan, BS, Clay J. Cockerell, MD, Kaveh Nezafati, MD, Joseph Susa, DO; Dallas, TX, Fort Worth, TX
History: A 71-year-old female who was has been on pembrolizumab for metastatic non-small cell lung carcinoma for approximately 1 year developed a 6-week duration of a generalized cutaneous eruption.
Physical Exam: Involving the chest and lower legs, there were several ill-defined erythematous and focally scaly macules, papules, patches and thin plaques. Involving the scalp, there were a few erythematous plaques with thick micaceous scale.
Histopathology: Biopsies from her scalp and chest revealed mounds of parakeratosis containing neutrophils overlying moderate psoriasiform hyperplasia with a moderately dense lymphoplasmacytic infiltrate in the dermis. A PAS stain was negative for hyphae and Treponema pallidum stain did not reveal spirochetes.
Clinical Course: The patient is currently being treated with oral methotrexate and topical steroids with significant improvement while continuing pembrolizumab therapy for her metastatic lung carcinoma.
Diagnosis: Psoriasiform eruption associated with pembrolizumab therapy
Points of Emphasis: Pembrolizumab is a monoclonal antibody used to treat metastatic or unresectable malignancies by preventing interaction with PD-1L on tumor cells with PD-1 on T cells, thereby boosting host T-cell activity. Numerous immune-mediated adverse effects have been reported with cutaneous involvement occurring in approximately 40% of treated patients(3). There is a wide range of cutaneous reactions including morbilliform/urticarial eruptions, pruritus, bullous pemphigoid, sarcoidosis, vitiligo/hypopigmentation, alopecia, eczematous, psoriasiform, and lichenoid eruptions. Reactivated T-cells is thought to be the underlying cause of the cutaneous adverse effects, however the exact mechanism is unknown. Studies show that the median interval between pembrolizumab initiation and cutaneous skin eruptions to be between 5-9 weeks after starting pembrolizumab, with the latest onset being around 24 months(3).
Our patient presented with a generalized eruption characterized by scattered, well-demarcated, erythematous scaly plaques on the scalp, chest, trunk, and extremities after approximately 1 year of treatment with pembrolizumab. A biopsy from the chest revealed psoriasiform epidermal hyperplasia with marked parakeratosis with neutrophils in the stratum corneum. Within the dermis, there was a moderately dense, somewhat band-like lymphoplasmacytic infiltrate with focal vacuolar change and scattered lymphocyte exocytosis within the epidermis. The reported histologic findings of cutaneous adverse events secondary to PD-1 inhibitors are varied and include what is described in this case(4,5). A scalp biopsy revealed marked parakeratosis encasing hair shafts correlating with tinea amiantacea clinically. The patient is currently being treated with oral methotrexate and topical steroids with significant improvement while continuing pembrolizumab therapy for her metastatic lung carcinoma.
In the setting of immune-related cutaneous adverse effects, it is thought that this may represent successful reactivation of T-cells and could theoretically correlate with better treatment response and longer progression-free intervals(3,6).
In conclusion, we present this interesting case with striking psoriasiform features, both clinically and histologically, in the setting of pembrolizumab treatment. It is important for clinicians to be aware of the possibility of immune-related cutaneous adverse effects of cancer immunotherapies that are being used more frequently. It is also important to realize that the timing of adverse effects may not be what is typically expected and may occur several months to years after initiation of therapy.
References: 1. ClinicalTrials.gov. Merck Sharp & Dohme Corp. 2013, Dec 5 . Identifier JAPIC-CTI, Study of Pembrolizumab (MK-3475) in Participants With Advanced Non-small Cell Lung Cancer (MK-3475-025/KEYNOTE-025); 2015, Dec 5. Available from: https://clinicaltrials.gov/ct2/show/results/NCT02007070?sect=X40156#oth 2. Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, Welch DR. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst1996; 88:1731-1737 3. Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab Cutaneous Adverse Events and Their Association With Disease Progression. JAMA Dermatol. 2015;151(11):1206–1212. doi:10.1001/jamadermatol.2015.1916 4. Shen J, Chang J, Mendenhall M, Cherry G, Goldman JW, Kulkarni RP. Diverse cutaneous adverse eruptions caused by anti-programmed cell death-1 (PD-1) and anti-programmed cell death ligand-1 (PD-L1) immunotherapies: clinical features and management. Therapeutic Advances in Medical Oncology. 2018;10:1758834017751634. doi:10.1177/1758834017751634.





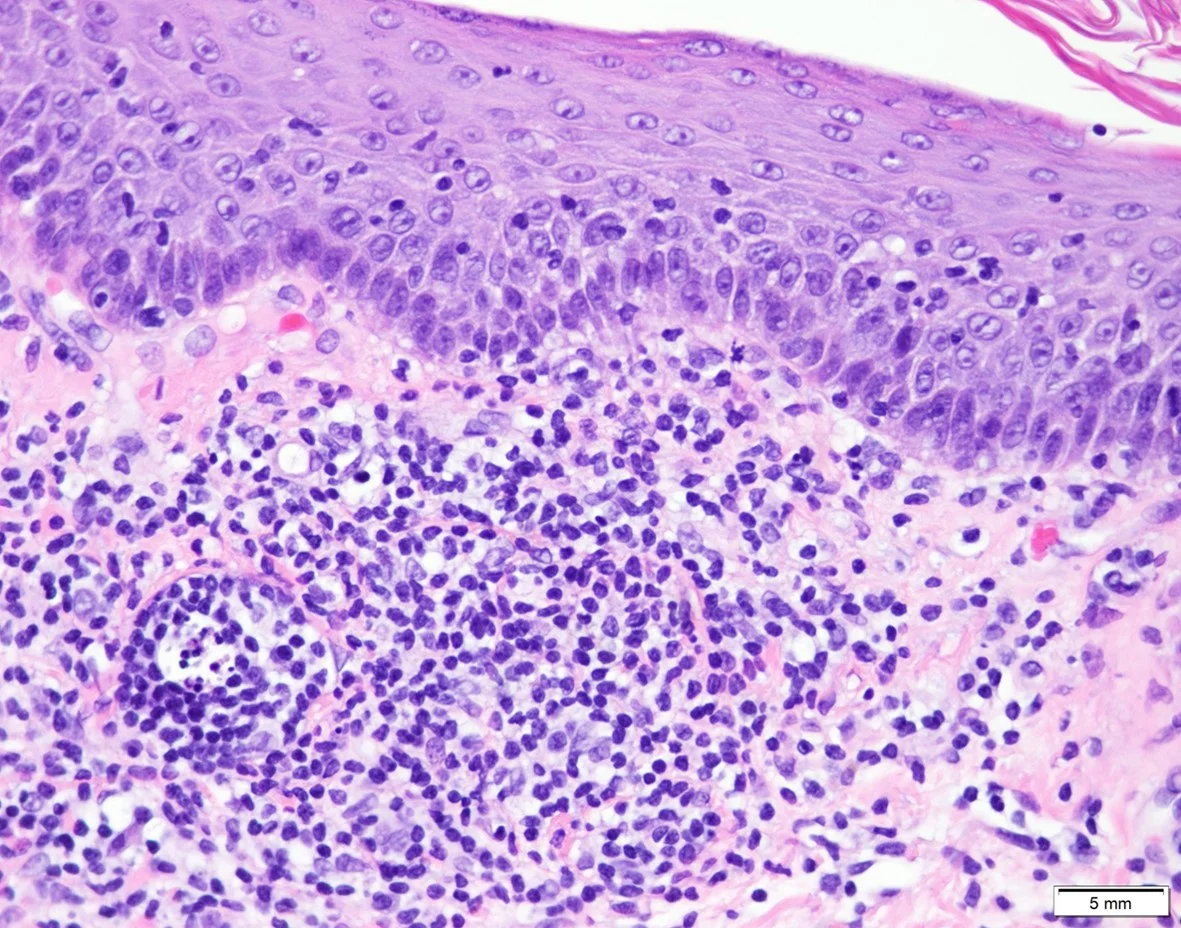

Case 25: An Unusual Case of Multicentric Reticulohistiocytosis
Caregivers: Travis Vandergriff, MD; Jennifer Day, MD; Jennifer Gill, MD, PhD; Dallas, TX
History: A 79 year old Hispanic woman with a history of hypertension, diabetes, and renal insufficiency presented to the hospital with complaints of new onset melena and hematemesis. She was noted to have a one year history of a rash which started on her back and later spread to involve the arms, legs, and chest. She had some pruritus but no other symptoms, and she had not attempted any treatments other than cetirizine. She had a history of mild arthritis.
Physical Examination: There were pink dermal papules coalescing into plaques on the chest and back. Lichenified erythematous plaques were found on the elbows. Around the first and second fingers, there were periungual skin-colored to pink papules. There was also abnormality of the knees with swelling and joint dystrophy.
Laboratory Data: Significant anemia with a Hgb 6.3
Histopathology: A punch biopsy from a papule on the back revealed a dense dermal infiltrate of epithelioid histiocytes with amphophilic “ground glass” cytoplasm in a nodular configuration.
Clinical Course: The patient was admitted for an upper GI bleed, and an EGD revealed a ulcerated mass in the antrum of the stomach. A biopsy of this mass showed changes of chronic reactive gastropathy as may be seen near an ulcer, but no neoplasm was noted. Her bleeding stabilized with proton pump inhibitors. A repeat EGD is planned for the future, with probable re-biopsy of the antral mass
Diagnosis: Multicentric reticulohistiocytosis
Points of Emphasis: Multicentric reticulohistiocytosis is a mucocutaneous papulo-nodular eruption formed by nodular accumulations of reticulohistiocytes in the skin. When papules coalesce into plaques, a cobblestone appearance can be seen. The papules tend to predominate around joint spaces and classically present with a beaded appearance in a periungual distribution. Reticulohistocytes are epitheiloid histiocytes with abundant amphophilic cytoplasm having a ground-glass texture. Such histiocytes may be seen in isolated/solitary lesions (reticulohistiocytoma) or in diffuse eruptions (diffuse cutaneous reticulohistiocytosis). In the diffuse form, patients often develop a destructive arthropathy. Solid internal malignancies are found in association in approximately 20% of cases.
In this case, the diffuseness of the process was striking and given the extent of the process, many of the areas appeared more like a papulosquamous disorder rather than a widespread granulomatous process. While she had evidence of large joint involvement, she did not have the usual mutilating, destructive arthropathy that often involves smaller joints such as those of the hand.
References: 1. Kumar B, Singh N, Rahnama-Moghadam S, Wanat KA, Ijdo JW, Werth VP. Multicentric Reticulohistiocytosis: A Multicenter Case Series and Review of Literature. J Clin Rheumatol 2018;24:45-49. 2. Selmi C, Greenspan A, Huntley A, Gershwin ME. Multicentric reticulohistiocytosis: a critical review. Curr Rheumatol Rep. 2015;17:511.














Case 26: Cutaneous Rosai-Dorfman Disease
Caregiver: Soham Chaudhari, DO; McAllen, TX
History: A 75-year-old Hispanic female with past medical history of basal cell carcinoma, treated with Mohs micrographic surgery previously, and hypertension presented to a South Texas dermatology clinic for evaluation of two “bumps” that had appeared within the last six months. She denied any symptoms from these lesions. Review of systems was negative for malaise, weight loss, night sweats, joint pain, or recent travel. She denied previous exposure to tobacco, alcohol, or drugs.
Physical Examination: A 0.9 cm x 0.9 cm erythematous nodule was noted on the right inferior eyebrow. No epidermal changes were appreciated; however, a central area of yellow debris was visualized. A similar appearing 0.5 cm x 0.5 cm papule without central yellow debris was present on the left lateral orbital rim. No cervical lymphadenopathy was appreciated on palpation.
Laboratory Data: CBC was unremarkable except for a Hgb level of 10 g/dL.
Histopathology: Within the dermis, there is a nodular infiltrate composed of lymphocytes, histiocytes, and plasma cells. Most of the histiocytes have abundant, clear to faint pink cytoplasm, and there is evidence of emperipolesis. GMS and AFB stains are negative for fungi and acid fast organisms, respectively. Immunohistochemical stains for CD1a are negative, while staining for S100 protein reveal decoration of the histiocytes.
Clinical Course: The patient was referred to an oncologist and no evidence of extracutaneous involvement was found.
Diagnosis: Cutaneous Rosai-Dorfman Disease
Points of Emphasis: Rosai-Dorfman disease is an uncommon histiocytic proliferative disorder. Although painless bilateral cervical lymphadenopathy is characteristic, extranodal sites, including the skin, can be the sole manifestation of the disease. The most commonly reported sites of extranodal involvement are the skin and soft tissue, which usually present as well-defined papules to palpable masses, with or without satellite lesions. In the lymph nodes and the skin, emperipolesis is a common histologic feature. The lesional histiocytes express both CD68 and S-100 protein while negative for CD1a, which helps differentiate this condition from Langerhans cell histiocytosis and Erdheim-Chester disease. Its clinical course is self-limiting, and simple excision is usually curative.
References: 1. Abla, O., E. Jacobsen, J. Picarsic, Z. Krenova, R. Jaffe, J. F. Emile, B. H. Durham, et al. "Consensus Recommendations for the Diagnosis and Clinical Management of Rosai-Dorfman-Destombes Disease." Blood 131, no. 26 (Jun 28 2018): 2877-90. 2. Dalia, S., E. Sagatys, L. Sokol, and T. Kubal. "Rosai-Dorfman Disease: Tumor Biology, Clinical Features, Pathology, and Treatment." Cancer Control 21, no. 4 (Oct 2014): 322-7. 3. Mantilla, J. G., S. Goldberg-Stein, and Y. Wang. "Extranodal Rosai-Dorfman Disease: Clinicopathologic Series of 10 Patients with Radiologic Correlation and Review of the Literature." Am J Clin Pathol 145, no. 2 (Feb 2016): 211-21.













Case 27: Erythromelanosis Follicularis Faciei
Caregivers: Amreen Sitabkhan, MD; Chaney Turney, MS4, Patricia Miller, MD;
History: A 23-year-old woman with a past medical history of Tetralogy of Fallot status post valve replacement and pacemaker installation presents to clinic with a rash isolated to her face, chest, and back that has persisted for months. She describes the rash as rough and denies pruritus as well as any associated symptoms. The patient has had no period of complete resolution of the rash since it began, and she notes no exacerbating nor alleviating factors. She endorses an allergy to sulfa drugs, and the patient is a lifetime non-smoker. She has not yet tried any treatments for her condition, and she takes no medications otherwise.
Physical Examination: Diffuse patches of follicular hyperkeratosis and scale are noted on the patient’s face, including forehead, chin, and bilateral cheeks. Follicular hyperkeratosis and overlying scale are also seen on her upper chest and back.
Histopathology: Sections show parakeratosis surrounding dilated follicular ostia. There is follicular distortion along with hyperkeratosis and plugging, and the hairs are surrounded by a sparse lymphohistiocytic inflammatory infiltrate. There is subtle patchy increase in pigment within the keratinocytes in the lower half of the epidermis, and there is an increased number of dilated blood vessels in the upper dermis.
Clinical Course: Patient has yet to return to clinic since receiving the biopsy results. Possible treatment modalities and management strategies will be discussed at follow up.
Diagnosis: Erythromelanosis follicularis faciei
Points of Emphasis: Erythromelanosis follicularis faciei is a rare condition characterized by follicular plugging, erythema, and hyperpigmentation of the facial skin. Symptoms most commonly develop during young adulthood and there tends to be a slight predilection for males, however the etiology of the condition remains unclear. The majority of reported cases are sporadic, but the possibility of an autosomal recessive inheritance pattern has been suggested. Often, associated keratosis pilaris is present on the trunk and extremities. Based on the clinical findings, the differential diagnosis includes keratosis pilaris rubra, poikiloderma of Civatte, Riehl’s melanosis, and pigmented peribuccal erythrosis of Brocq.1 Various treatment modalities have been described with limited success, with topical retinoids, ammonium lactate lotion, metronidazole, and hydroquinone being the most frequently cited anecdotal trials.2
References: 1. Warren, F. M., & Davis, L. S. (1995). Erythromelanosis follicularis faciei in women. Journal of the American Academy of Dermatology, 32(5), 863-866. Retrieved from https://www.jaad.org/article/0190-9622(95)91548-6/fulltext. 2. Al Hawsawi, K., & Aljuhani, O. (2015). Erythromelanosis Follicularis Faciei: A Case Report and Review of the Literature. Case Reports in Dermatology. Retrieved from https://www.karger.com/Article/FullText/442343.

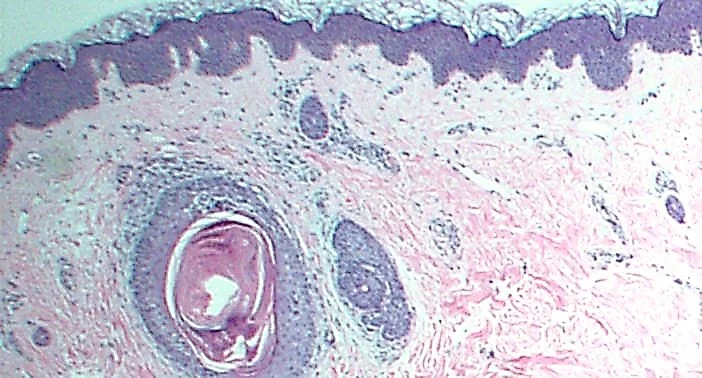


Case 28: Juxta-Clavicular Beaded Lines
Caregivers: Aatika Malik MD1, Joseph Susa DO2, Steve Weis MD2, Clay Cockerell M.D2; Philadelphia, PA1, Dallas, TX2
History: A 66 year old Native American male smoker presented to the dermatology clinic with complaint of spots started on his neck 18 to 20 years ago. The lesions were spreading down his trunk and stopped below his breasts. It was also spreading down his shoulders and his arms. He denied any itching, burning or pain. He was seen by two dermatologists and treated for eczema with triamcinolone for three years and betamethasone for one month respectively. He denied any improvement. He had a past medical history of COPD and seasonal allergies. He had been taking inhaled steroids for years. He had worked outside all his life and had extensive sun exposure.
Physical Examination: Physical examination revealed sun damaged skin and solar purpura on his arms. Multiple yellow to flesh colored papules ranging from 1-2 mm in size involving neck, chest and bilateral upper extremities were noted. Nipples were spared.
Histopathology: The histological examination of a shave biopsy showed an irregular epidermis. There were a number of sebaceous lobules with a sebaceous follicle and a few leukocytes in the dermis. The appearance was that of a benign sebaceous hyperplasia. The findings were relatively subtle although in the context of clinical photographs, they were compatible with the diagnosis of juxta-clavicular beaded lines.
Clinical Course: Topical steroids were prescribed with minimal change.
Diagnosis: Juxta-clavicular beaded lines.
Points of Emphasis: Juxta-clavicular beaded lines is a rarely described variant of the normal skin of the neck and clavicular area. Small skin colored to white to yellow brown papules ranging from 1-2 mm in diameter are oriented along Langer’s lines of tension of the neck, clavicular area, and less commonly the axillae. These papules occasionally coalesce, however the pattern remains as parallel stippled bands. The condition is asymptomatic and is not known to be associated with any underlying conditions. Clinicians unaware of the diagnosis may confuse it with eczema as happened in our case. It has been described in patients receiving systemic corticosteroids. Steroid therapy results in skin atrophy leading to more pronounced lesion. Other differential diagnoses with similar lesion include acanthosis nigricans, papular mucinosis and pseudoxanthoma elasticum. The skin-toned to white color in our case helped us to differentiate it from acanthosis nigricans which is typically a velvety textured hyperpigmented color, and papular mucinosis which is typically a pale red to yellow color. Histologically, papular mucinosis has abundant acid mucopolysaccharides present in the dermis and an increased number of fibroblasts. Pseudoxanthoma elasticum is more yellow in color with histologically evident wider elastic fibers and surrounding calcium deposits. Pseudoxanthoma elasticum-like papillary elastolysis clinically appears similar to pseudoxanthoma elasticum but is found more frequently in elder women and histologically shows a band like loss of elastic tissue in the papillary dermis.
References: 1. Woldow, Adam B., Laura D. Houk, and Faramarz H. Samie. "Juxtaclavicular beaded lines: A presentation of sebaceous gland hyperplasia." Dermatology online journal 15.4 (2009). 2. Lee, Sang Eun, You Chan Kim, and Seung Hun Lee. "Juxta‐clavicular beaded lines." The Journal of Dermatology 34.6 (2007): 407-409. 3. Franco, Gennaro, et al. "Juxta‐clavicular beaded lines." Australasian Journal of Dermatology 47.3 (2006): 204-205.




Case 29A: Atypical (Persistent) Eruption of Adult-onset Still’s Disease
Caregivers: Leah Persad, DO; Kevin Honan, BS; Travis Vandergriff, MD Dallas, TX
History: A 35-year-old woman with a past medical history of hypertension presenting with a two month duration of persistent and pruritic eruption along with intermittent high fevers, arthritis of both knees with associated swelling and morning stiffness, nausea, vomiting, and weight loss.
Physical Examination: Physical exam was notable for focally linear and rippled hyperpigmented papules and plaques on the chest, back, and proximal medial thighs.
Laboratory Data: Leukocytosis (>80% neutrophils), hemolytic anemia, elevated ferritin (10,000, downtrended to 3756 at the end of hospitalization), elevated creatinine (1.49), elevated liver function tests (AST: 201, ALT: 1061, both trended to normal values at the end of hospitalization, ANA (-), RF (-)
Histopathology: A punch biopsy of the chest was performed and revealed a superficial perivascular infiltrate consisting of lymphocytes and neutrophils with focal vacuolar alteration and scattered necrotic keratinocytes which predominate in the upper levels of the epidermis.
Diagnosis: The clinical and histopathological findings were most consistent with adult-onset Still’s disease with an atypical (persistent) cutaneous eruption.
Clinical Course: The patient clinically improved on high-dose systemic steroids. Her course was complicated by biopsy-proven acute interstitial nephritis, unsure to be related to AOSD. She required 3 units of RBCs while inpatient. She was discharged on prednisone 80 mg daily. Her skin continued to improve and was discharged with oral diphenhydramine, hydroxyzine and topical triamcionlone 0.1% cream.
Points of Emphasis: Adult onset Still’s disease (AOSD) is a rare, systemic autoinflammatory disease that is characterized by intermittent high spiking fevers, arthritis and/or generalized arthralgias, and an evanescent, salmon-colored skin rash; this conglomerate of findings is in accordance with Yamaguchi et al and the Yamuaguchi criteria[1]. Labs are often significant for leukocytosis and hyperferritinemia[2]. The etiology is still unknown but studies have shown certain HLA predispositions[3]. The diagnosis of AOSD can often be delayed due to the nonspecificity of the symptoms as well as the fact that patients may sometimes present with atypical findings. Atypical cutaneous presentations in AOSD include persistent papules and plaques that is sometimes in a linear/reticular pattern, persistent urticarial rashes/urticarial-like eruptions, generalized non-pruritic erythema, and vesiculopustular eruptions. In approximately half of these atypical cutaneous lesions, the classical evanescent skin rash exists concurrently [5, 6]. Delayed diagnosis can increase morbidity and patients are at an increased risk for serious complications of untreated AOSD such as macrophage activation syndrome, DIC, TTP, and pulmonary arterial hypertension[4].
Our patient presented with some of the common systemic symptoms as well an atypical cutaneous eruption consisting of persistent, pruritic, linear/reticulated papules and plaques on the chest, back, and proximal medial thighs. A skin biopsy revealed focal vacuolar alteration and scattered necrotic keratinocytes that predominated in the upper levels of the epidermis. This histologic finding has been reported in the atypical (persistent) cutaneous eruptions in AOSD[5]. The constellation of clinical findings is relatively non-specific in AOSD, however the characteristic histology of the atypical cutaneous presentation was invaluable in making the diagnosis of AOSD in this patient.
Treatment tailored towards the control of atypical cutaneous lesions in AOSD consists of glucocorticoids, immunosuppressive drugs and, in refractory cases, biologic therapy (notably, TNF-alpha inhibitors, IL-1 antagonists)[7]. Glucocorticoid and antibiotics, in combination, have been reported to curb the initial flare of atypical cutaneous lesions but lack in ability to prevent relapse[6].
We present this interesting case in hopes that the findings will contribute to the existing literature of this rare disease and its atypical manifestations – in doing so, we aim to help expedite future diagnoses of AOSD, decreasing both morbidity and mortality in these patients.
References: 1. Yamaguchi, M., et al., Preliminary criteria for classification of adult Still's disease. J Rheumatol, 1992. 19(3): p. 424-30. 2. Meijvis, S.C., et al., Extremely high serum ferritin levels as diagnostic tool in adult-onset Still's disease. Neth J Med, 2007. 65(6): p. 212-4. 3. Pouchot, J., et al., Adult Still's disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore), 1991. 70(2): p. 118-36. 4. Efthimiou, P., S. Kadavath, and B. Mehta, Life-threatening complications of adult-onset Still's disease. Clin Rheumatol, 2014. 33(3): p. 305-14. 5. Fortna, R.R., et al., Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol, 2010. 37(9): p. 932-7. 6. Santa, E., et al., Clinical and histopathological features of cutaneous manifestations of adult-onset Still disease. J Cutan Pathol, 2017. 44(6): p. 591-595. 7. Gopalarathinam, R., et al., Adult Onset Still's Disease: A Review on Diagnostic Workup and Treatment Options. Case Rep Rheumatol, 2016. 2016: p. 6502373.






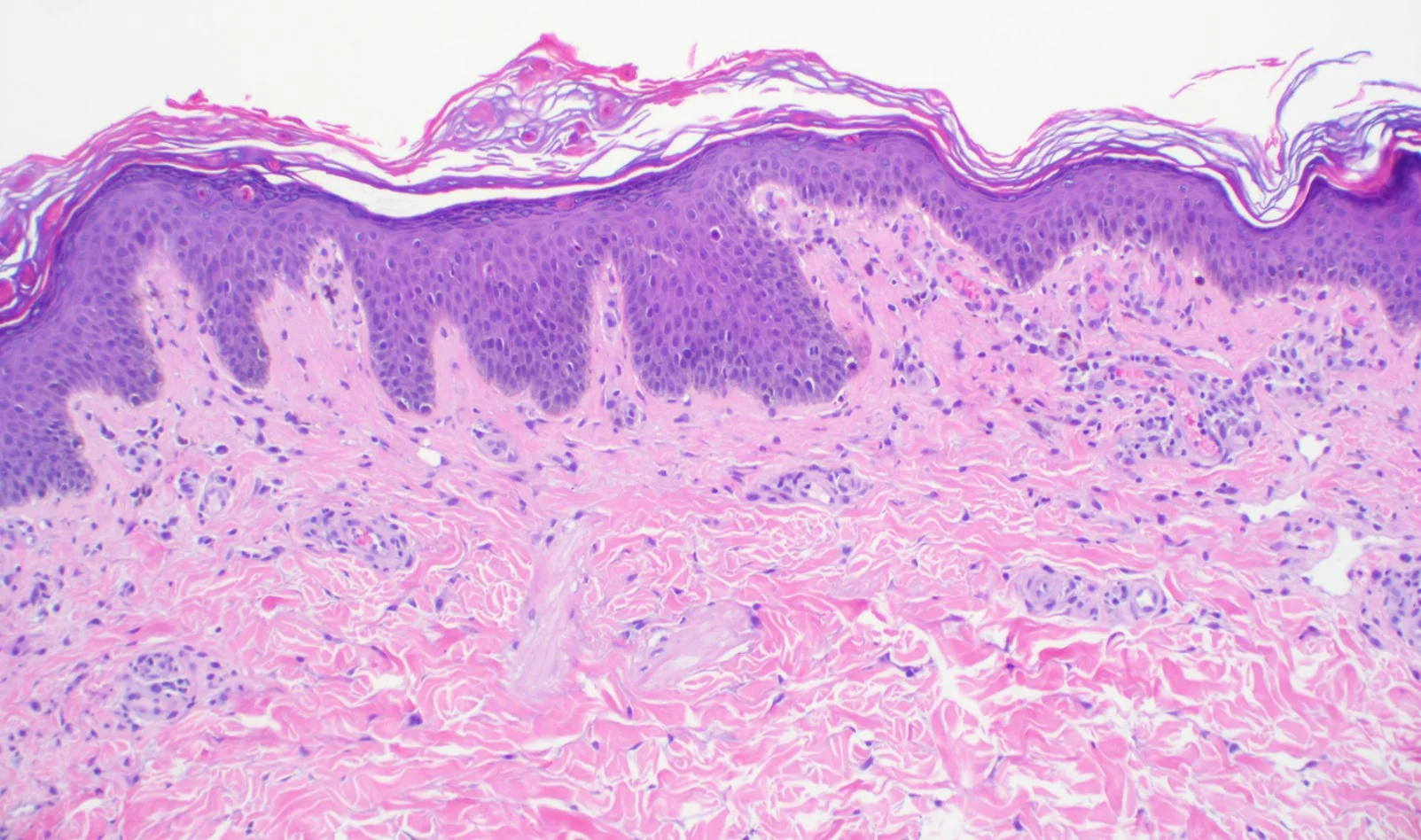




Case 29B: Adult Onset Still’s Disease with Macrophage Activation Syndrome
Caregivers: Travis Vandergriff, MD, Arturo Dominguez, MD, Etan Marks, DO, Dallas, TX
History: A 19 year old Caucasian female with depression was transferred from an outside hospital with weakness, fevers, and chills. She had leukocytosis, predominantly neutrophilic, with elevated band forms up to 24%. Her CRP, ESR and ferritin were elevated with a negative ANA and Rheumatoid factor. An infectious work up was negative. Rheumatology eventually diagnosed her with adult onset Still’s disease. Subsequently, she developed a chronic eruption on her legs.
Physical Examination: Physical exam was notable for atypical annular papules on her legs that would leave purpuric macules behind, concerning for urticarial vasculitis.
Laboratory Data: Very high ferritin and IL-18
Histopathology: A biopsy was performed that showed an inflammatory cell infiltrate of lymphocytes, histiocytes, abundant neutrophils, rare eosinophils and extravasated erythrocytes in the dermis. No fibrin was seen in vessel walls. However, in and around some of the deeper dermal vessels were histiocytes which had engulfed degenerative inflammatory cells including lymphocytes. Therefore, a diagnosis of urticaria with extravasated erythrocytes and hemophagocytosis was made.
Diagnosis: Adult onset Still’s disease with associated macrophage activating syndrome.
Clinical Course: She had an excellent response to the combination of canakinumab and tacrolimus and we have been able to taper her steroid without issue. Her ferritin is currently in the 200s. Her rash is improved, her myalgias and arthralgias (with the exception of her back pain) are improved, and she was able to be discharged.
Points of Emphasis: HLH is thought to occur because of widespread immune activation with proliferation of cytotoxic CD8+ T cells and macrophages which eventuate in hemophagocytosis. Early diagnosis and treatment are essential for preventing a suboptimal outcome when HLH occurs in this setting. A combination of therapies, including anakinra, systemic glucocorticoids, and cyclosporine can possibly improve clinical outcomes in these patients.
References: 1. Arneill M, Arneill R, Maiden N (2014) Macrophage Activation Syndrome Secondary to Adult Onset Still’s Disease- An Important Sepsis Mimic. JSM Clin Case Rep 2(2): 1022. 2. Eimear Savage, Tasheen Wazir, Mary Drake, Robert Cuthbert, Gary Wright; Fulminant myocarditis and macrophage activation syndrome secondary to adult-onset Still’s disease successfully treated with tocilizumab, Rheumatology, Volume 53, Issue 7, 1 July 2014, Pages 1352–1353,


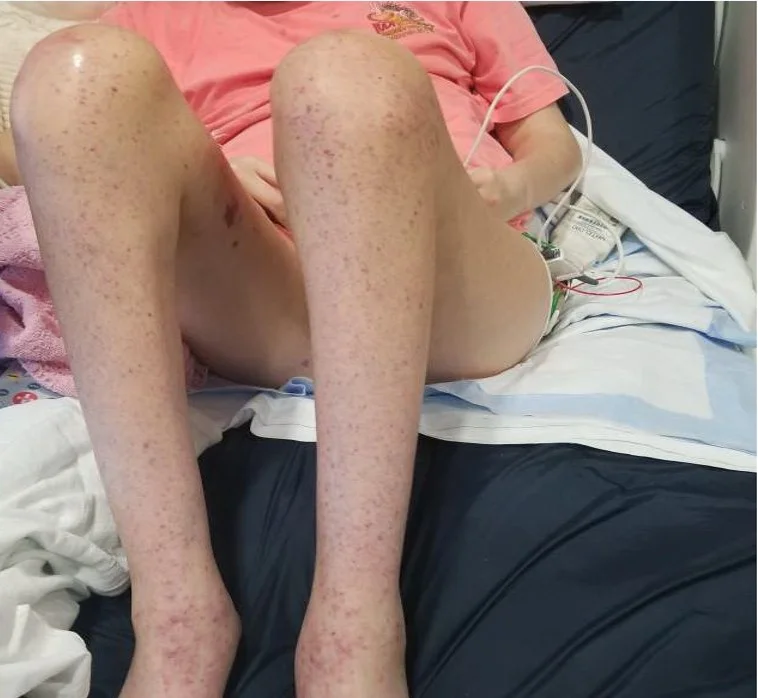





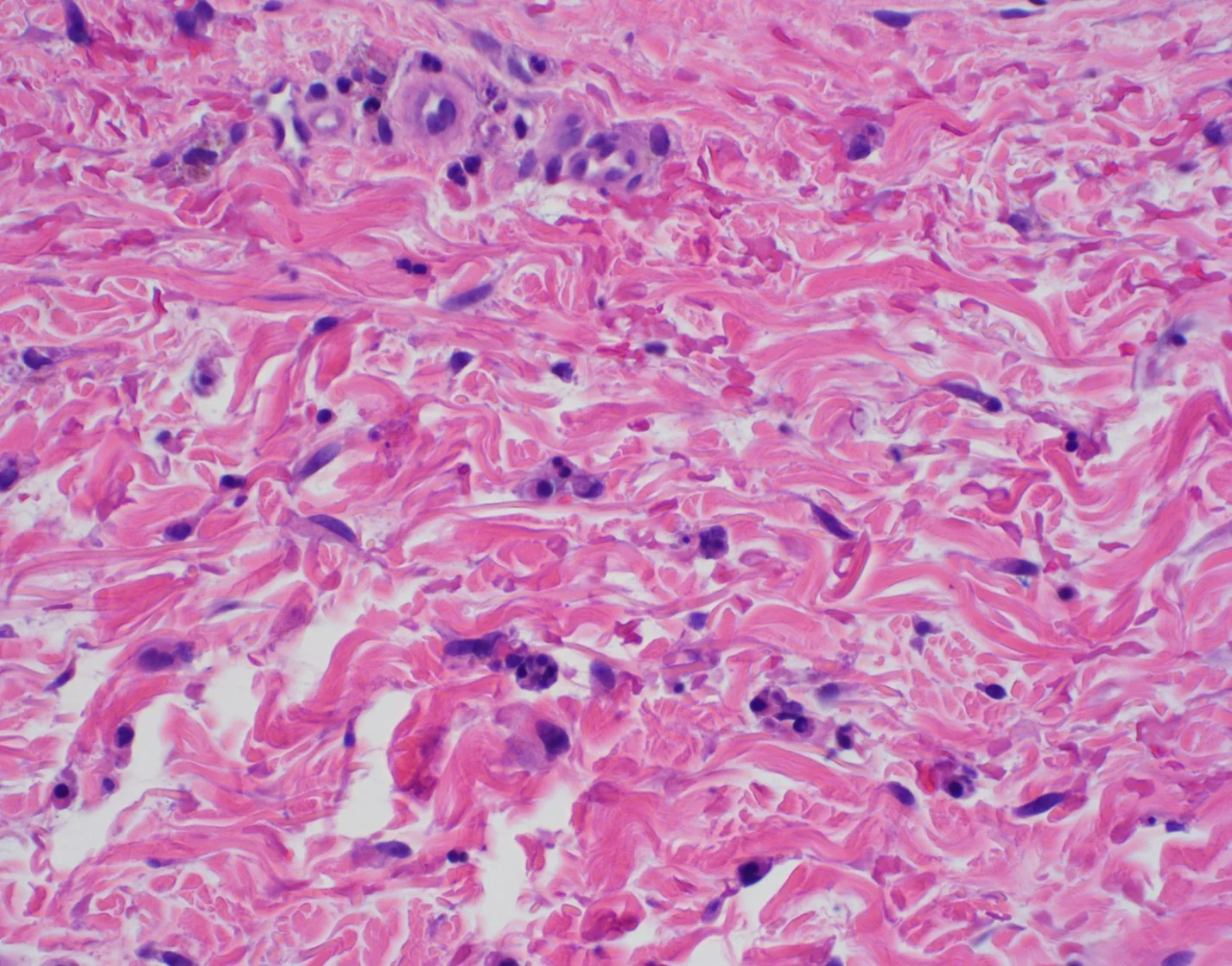

Case 30: Symmetrical Drug Related Intertriginous and Flexural Exanthema (SDRIFE)
Caregivers: Leah Persad, DO, Carolyn B. Lyde, MD, Clay J. Cockerell, MD and Joseph Susa, DO; Dallas, TX and Lewisville, TX
History: A 62-year-old woman with a history of hypertension was started on a new medication, Diltiazem ER, and developed a diffuse pruritic eruption 13 days later. Initially, the eruption was treated with an intramuscular injection of ceftriaxone and dexamethasone along with oral clindamycin, fluconazole, and oral hydroxyzine with topical mupirocin ointment.
Physical Exam: Involving the bilateral axillae, inguinal folds, trunk, and focally on the extremities are discrete and coalescing, focally round and targetoid erythematous to violaceous minimally scaling macules, patches, and thin plaques. There were no associated systemic symptoms.
Histopathology: Punch biopsies from the back revealed psoriasiform dermatitis with overlying parakeratosis and focal mounds of neutrophils in the stratum corneum. Involving the superficial dermis, there were a few scattered interstitial eosinophils, neutrophils, and extravasated red blood cells.
Clinical Course: The patient slowly improved after Diltiazem ER was discontinued.
Diagnosis: Urticarial drug reaction with psoriasiform features consistent with symmetric drug-related intertriginous and flexural exanthema.
Points of Emphasis: Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a form of systemic allergic dermatitis induced by a systemically administered drug. This eruption was previously called “baboon syndrome” as it commonly occurs on the buttocks as well as the intertriginous areas of the body. Beta-lactam antibiotics are the leading offending medication with case reports also including clarithromycin, doxycycline, metronidazole, infliximab, golimumab, and many more.
Clinically, the eruption is characterized by moderately ill-defined erythematous patches or thinly scaling plaques in intertriginous areas (axillae, groin, neck) as well as scattered on the trunk and extremities. Rarely, there may be pustulobullous lesions also. The eruption may develop within a few hours up to 2 weeks after the drug has been administered. The differential diagnosis includes a morbilliform drug eruption, inverse psoriasis, intertrigo, atopic dermatitis, and acute generalized exanthematous pustulosis. The clinical history of a possible offending medication is necessary to making this diagnosis.
Histopathologically, SDRIFE has been described to show non-specific inflammatory findings including mild spongiosis, focal vacuolar change, necrotic keratinocytes, a superficial perivascular lymphocytic infiltrate with interstitial dermal eosinophils and neutrophils.
It is thought that most of these reactions are a Type IV delayed hypersensitivity reaction mediated by cytotoxic T cells. In this interesting clinical case, the patient demonstrated the symmetric intertriginous eruption with focal areas resembling a fixed drug eruption clinically with mostly non-specific psoriasiform changes on histopathology.
Identifying and discontinuing the causative medication is imperative. Patch testing is possible but may have limited utility as there is limited absorption through the skin versus systemically and may not elicit a reaction. Controlled oral provocation testing has been thought to be useful in confirming the diagnosis. Along with discontinuation of the drug, oral anti-histamines and topical corticosteroids can be used.
References: 1. Nespoulous L, Matei I, Charissoux A, Bédane C, Assikar S. Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) associated with pristinamycin, secnidazole, and nefopam, with a review of the literature. Contact Dermatitis. 2018 Jul 30. doi: 10.1111/cod.13084 2. Labadie JG, Florek AG, Croitoru A, Liu W, Krunic AL. First case of symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) due to Berberine, an over-the-counter herbal glycemic control agent. Int J Dermatol. 2018 Sep;57(9):e68-e70 3. Ozkaya E, Babuna G. A challenging case: Symmetrical drug related intertriginous and flexural exanthem, fixed drug eruption, or both? Pediatr Dermatol. 2011 Nov-Dec;28(6):711-4 4. Dogru M, Ozmen S, Ginis T, Duman H, Bostanci I. Symmetrical drug-related intertriginous and flexural exanthema (baboon syndrome) induced by amoxicillin-clavulanate. Pediatr Dermatol. 2012 Nov-Dec;29(6):770-1











Case 31: Partially Erythrodermic Urticaria Pigmentosa
Caregivers: Andrew DeCrescenzo, MD, Leah Persad, DO, Clay J. Cockerell, MD, Samir Patel, MD, Mona Foad, MD; Dallas, TX, Cincinnati, OH
History: A 63-year-old female presented with a one-year history of waxing and waning pruritic erythematous to purpuric cutaneous eruption. Trials of oral prednisone and topical triamcinolone did not improve the pruritus. Despite her chronic pruritus, she never developed constitutional symptoms, GI symptoms, or bony pain.
Physical Exam: The patient was ambulatory and appeared well. Her skin showed small monomorphic reddish-brown macules and papules that formed focally confluent patches and plaques with much a deeper violaceous/purpuric hue on the thighs. The distal extremities were relatively spared.
Laboratory Data: Non-contributory; no elevated mast cell mediators noted
Histopathology: Punch biopsy from the left thigh showed sparse infiltration of mononuclear cells in the superficial dermis with features of mast cells with minimal epidermal change. Tryptase stain was diffusely and strongly positive.
Clinical Course: The patient was referred for bone marrow biopsy to exclude bone marrow involvement or an underlying mast cell leukemia.
Diagnosis: Urticaria pigmentosa with partially erythrodermic features
Points of Emphasis: Mast cell disease or mastocytosis is a disorder that has multiple phenotypes and can present at any age. The hallmark is mast cell proliferation, which may involve skin, internal organs, or both. The prototypical mutation is an activating mutation of KIT. In children, the disease is typically limited to only skin with resolution by adolescence; however, in adults, varying degrees of systemic involvement and a prolonged clinical course is typical.1
The most common childhood-onset mastocytosis is that of urticaria pigmentosa (UP), followed by solitary mastocytoma, and rarely diffuse cutaneous mastocytosis (DCM).2 The exam of childhood UP reveals small, monomorphic, tan-brown macules or papules as well as large polymorphic yellow-tan to brown plaques and nodules. Phenotypes with few clustered lesions on the trunk to uncountable lesions forming confluent plaques that typically favor the trunk and proximal extremities and spares the mid-face and palmoplantar surfaces highlights the varied presentation of this entity. On the other hand, solitary mastocytoma appears with a yellow-tan to brown nodule or plaque with a leathery texture that favors the distal extremities. The rare DCM presents in the neonatal period with thickened leathery skin with variable hyperpigmentation—it is truly generalized and should be distinguished from a confluence of individual lesions.
Above highlights three of the most commonly seen types of childhood-onset mastocytosis; however, there are atypical presentations that appear with ill-defined tan patches with telangiectasia, brown nodules, or with yellow superficial nodules simulating xanthomas. It is important to retain mast cell disease on the differential diagnosis of children with truncal papules and plaques of the yellow-brown palette.
The most common presentation of adult-onset mastocytosis appears with small, monomorphic, reddish-brown macules and papules favoring the trunk and proximal extremities while sparing the central face and palmoplantar surfaces. This presentation is similar to the childhood presentation of urticaria pigmentosa, although typically does not have the yellow-brown features of the pediatric disease. Our patient presented with this form of mastocytosis, however the confluence of her macules and papules was striking as it focally resembled a near erythrodermis state. Adult cutaneous mastocytosis can also present with macules and patches comprised of telangiectasia without the hyperpigmentation common to mast cell disease called telangiectasia macularis eruptiva perstans (TMEP). Uncommonly, adult disease presents with yellow-tan to red-brown nodules—a hallmark of systemic mastocytosis.
In this case, the diagnosis was unusual as the lesions were so coalescent as to produce a presentation that simulated an erythrodermic process in some areas.
The confirmation of cutaneous mastocytosis with histopathology should prompt an evaluation for systemic involvement—typically of the bones and bone marrow. The pre-test probability for bone marrow involvement is essentially 0% in the pediatric population, and routine bone marrow evaluation does not affect subsequent therapy; however, adults with extensive cutaneous disease will invariably have some degree of bone marrow involvement.3,4
The treatment of cutaneous mastocytosis hinges on avoiding triggers of mast cell degranulation such as aspirin, narcotics, alcohol, and anesthetics. Symptomatic relief for pruritus with H1 antagonists and gastrointestinal symptoms with H2 antagonists align with the pathophysiology of mast cell mediated histaminergic disease. Conversely, treatment of systemic mastocytosis is highly individualized and is guided by genetic analysis and is beyond the scope of this summary.
References: 1. Akin C, Valent P. Diagnostic criteria and classification of mastocytosis in 2014. Immunol Allergy Clin North Am. 2014;34(2):207-218. 2. Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol. 2011;12(4):259-270. 3. Pardanani A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am J Hematol. 2016;91(11):1146-1159. 4. Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137(1):35-45.








Case 32: Primary Cutaneous Nocardiosis
Caregivers: Mark Thieberg, MD, Etan Marks, DO, Clay J. Cockerell, MD; Dallas, TX
History: A 71 year old healthy male presented with an edematous and erythematous eruption with areas of pustules and erosions. The clinical differential diagnosis was cellulitis caused by a staphylococcus or streptococcus, herpes zoster virus infection or possibly pyoderma gangrenosum.
Physical Examination: Involving the right posterior calf are several closely set several centimeter erythematous, focally purulent and necrotic ulcerated nodules with surrounding warm, tender, edematous erythema.
Histopathology: Diffuse suppurative inflammation in the dermis with prominent papillary dermal edema. PAS, Gram and Fite stains were negative for infectious organisms.
Clinical Course: Based on the morphology of the lesions and the histologic findings of prominent suppuration and negative special stains for microorganisms, systemic corticosteroids were initiated. Shortly after initiation of the steroids, a culture was reported as being positive for Nocardia. Subsequently, the steroids were discontinued and IV Bactrim was started. The patient responded well to therapy and was discharged on oral Bactrim.
Laboratory Data: Cultures positive for Nocardia and was further classified as Nocardia brasiliensis/vulneris by DNA sequencing.
Diagnosis: Cutaneous Nocardiosis
Points of Emphasis: Cutaneous nocardiosis is an uncommon infectious disease that presents as either a primary cutaneous infection or as part of a disseminated pulmonary nocardiosis. The main route of infection is direct inoculation through the skin and, less commonly, direct inhalation of the bacteria which is then disseminated to the skin.
Nocardia usually occurs in immunoompromised patients but in rare cases it may occur in immunocompetent patients as well. There are several species of Nocardia that can infect humans, with N. asteroides being the most common, often manifesting as a pulmonary or CNS infection and, rarely occuring as a disseminated disease which can manifest in the skin. Primary cutaneous nocardiosis, which is extremely rare, has been reported to be caused by N. brasiliensis as well as N. asteroides. Clinically, cutaneous nocardiosis appears as either a lymphocutaneus syndrome, superficial skin infection (pustules, pyroderma, abscess, ulcers, granulomas, or cellulitis) or a mycetoma.
The diagnosis of nocardiosis relies on isolation of organisms by smear and by culture from skin biopsies. Growth is usually seen after 2–7 days. Optimal antimicrobial regimens have not been firmly established for the treatment of nocardiosis especially since the organism displays variability with regard to in vitro antimicrobial susceptibility patterns. However, cure rates for patients with primary cutaneous nocardiosis are almost 100%.
The organisms stain weakly Acid Fast positively and appear as small coccobacillary forms. While there may be “sulfur granules” in tissue, there may be few organisms in tissue as in this case which caused the clinician to consider the diagnosis of a neutrophilic dermatosis and initiate treatment with systemic steroids which could have caused the process to spread. This case emphasizes the importance of performing tissue cultures in unusual cases to exclude the diagnosis of an occult infection.
References: 1. Dodiuk-Gad R, Cohen E, Ziv M, et al. Cutaneous nocardiosis: report of two cases and review of the literature. Int J Dermatol. 2010;49(12):1380–1385. 2. Satterwhite TK, Wallace RJ. Primary cutaneous nocardiosis. JAMA. 1979;242(4):333–336.





Case 33: Dysmorphic Trichophyton rubrum Infection Simulating Blastomycosis
Caregivers: Richard Hope, MD, Etan Marks, DO, Clay J. Cockerell MD; Lubbock, TX and Dallas, TX
History: A 60 year old male with a recent history of kidney transplantation, 9 months previously presented to the dermatology clinic concerned about a rash present on his arms, abdomen, back and legs which had been present for 6 months. Notably, the patient states he had shingles on his right foot after the transplant which was treated. Soon thereafter, the rash appeared.
Physical Exam: Scattered erythematous papules on his chest and right thigh. The right calf had several plaques with erythematous scale and annular configuration and his right ankle had several erythematous firm nodules. A punch biopsy was performed.
Histopathology: There was a diffuse suppurative granulomatous dermatitis with pseudocarcinomatous hyperplasia. Within the zones of suppuration and within multinucleated histiocytes, broad based budding fungal structures were visualized. This was confirmed with a PAS stain. The morphology was thought to be consistent with blastomycosis. A tissue culture was performed which grew Trichophyton rubrum. Becauese of the unusual morphology, tissue was sent to the CDC for DNA extraction and confirmation and to exclude the diagnosis of blastomycosis that might have been missed on culture because of overgrowth with T. rubrum.
The test from the Centers for Disease Control failed to extract DNA revealed hyphal elements rather than broad based budding yeasts. Given the results of the cultures, the diagnosis of a dysmorphic variant of Trichophyton simulating blastomycosis was made.
Laboratory Data: Cultures grew Trichophyton rubrum
Clinical Course: The patient was treated with itraconazole for 1 year and the lesions improved. A subsequent biopsy was performed to assess if any residual fungus remained in the tissue which was negative revealing only hypertrophic scar.
Diagnosis: Dysmorphic Trichophyton rubrum infection simulating blastomycosis
Points of Emphasis: Dermatophyte fungi can appear pleomorphic in tissue and demonstrate unusual morphology that may simulate other fungal organisms. This is thought to be an adaptive response to the harsh alkaline environment of the dermis. Similar to our case, 2 prior reports showed the fungus mimicked blastomycosis presenting with round spore-like shapes and appeared to have broad based budding. It is worth noting that in specimens with few organisms, normal hyphae cut in cross-section can be mistaken for round yeasts with budding. Clinicians and dermatopathologists should be alert to the variability of presentation of invasive dermatophytes, especially in immunocompromised hosts, and the importance of culture or molecular testing in addition to histology to establish the diagnosis.
References: 1. Smith KJ, Welsh M, Skelton H. Trichophyton rubrum showing deep dermal invasion directly from the epidermis in immunosuppressed patients. Br J Dermatol. 2001;145(2):344-348. 2. Akman A, Savas B, Uguz A, et al. Invasive Trichophyton rubrum infection resembling blastomycosis in a patient with altered immune status during the course of chronic superficial Trichophyton rubrum infection. Eur J Dermatol. 2007;17(5):452-453. 3. Talebi LF, Shinohara MM. Invasive Trichophyton rubrum mimicking blastomycosis in a patient with solid organ transplant. Journal of Cutaneous Pathology. 2017;44(9):798-800.








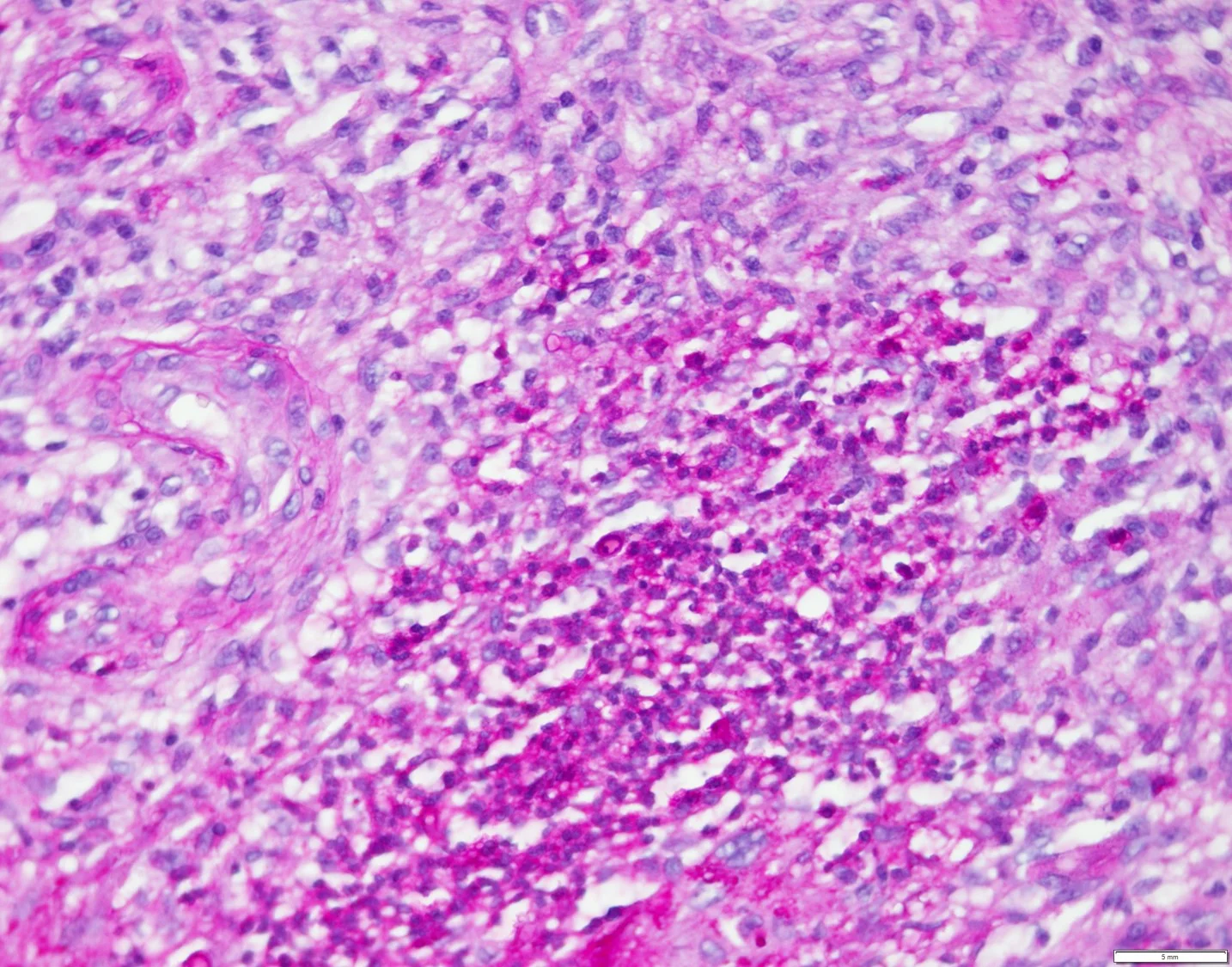
Case 34: Disseminated Coccidioidomycosis in a Young Boy
Caregivers: Kavina Patel, BS; Zachary Gillooly, MD; Sandra Osswald, MD; San Antonio, TX
History: A 15-year-old healthy Middle Eastern male originally presented to the hospital with a four-week history of ongoing fevers to 103°F, dyspnea, pleuritic chest pain, nonproductive cough, and rash concerning for erythema nodosum. He had recently moved to Texas from Arizona. Chest CT scan showed pulmonary nodules and hilar lymphadenopathy. He was diagnosed with pulmonary coccidioidomycosis. He was not treated at the time due to immunocompetency and lack of extrapulmonary signs or symptoms. The patient re-presented to the hospital two months later with back and bilateral foot pain as well as two solitary skin lesions.
Physical Examination: The patient had a single 1cm violaceous, soft, crusted papule with central erosion on the left temporal scalp and a single 2cm violaceous, slightly indurated plaque with central erosion and sinus tract on the left lower back (see photographs). Remainder of the skin exam was unremarkable.
Laboratory Data: Cocci enzyme immunoassay IgM: 4.0 IV (normal: <0.9 IV); cocci enzyme immunoassay IgG: 2.7 IV (normal: <0.9 IV); complement fixation: 1:2 (normal: less than 1:2); white blood cell: 14.5 x 109/L (normal: 4.5-11 x 109/L).
Histopathology: Histopathologic examination revealed non-caseating granulomatous inflammation with multinucleated giant cells and pseudoepitheliomatous hyperplasia with neutrophils. On higher magnification, spherules (sporangia) with endospores (sporangiospores) were present suggestive of Coccidioides.
Clinical Course: Fungal culture from skin biopsy was positive for cocci. An MRI of both feet demonstrated changes consistent with disseminated coccidioidomycosis and osteomyelitis. A bone biopsy of the L2 vertebra was positive for cocci, and a lumbar puncture resulted in a negative CSF fungal culture. The patient was treated with a 14-day course of amphotericin B along with fluconazole, which may be required for lifelong maintenance treatment.
Diagnosis: Disseminated coccidioidomycosis infection of the skin and bone
Points of Emphasis: Cutaneous manifestations of coccidioidomycosis include a secondary skin infection due to dissemination (as seen in this patient), primary cutaneous inoculation, or a reactive pulmonary exanthem commonly manifesting as erythema nodosum, toxic exanthem, or erythema multiforme-like lesions.1,2 Skin lesions secondary to disseminated coccidioidal infection include papules, nodules, verrucous plaques, pustules, abscesses, and sinus tracts and are most commonly located on the scalp, face, neck, and chest. In disseminated infections, Coccidioides is observed microscopically and grows in culture from biopsy.
Uncomplicated primary pulmonary coccidioidomycosis in an immunocompetent individual does not warrant antifungal treatment, and cases usually resolve spontaneously. However, more severe manifestations of coccidioidomycosis require treatment with antifungal agents such as amphotericin B deoxycholate (AmB), fluconazole, or itraconazole.3 Treatment is generally indicated when the patient becomes symptomatic or has certain risk factors. According to the 2016 Infectious Diseases Society of America (IDSA) cocci treatment guidelines, for asymptomatic immunocompetent patients with primary pulmonary cocci or a coccidioidal cavity, no antifungal treatment is recommended. The presence of pulmonary infiltrates in both lungs or more than 50% of one lung, hilar adenopathy, anticoccidioidal complement fixation titer > 1:16, and symptoms lasting more than two months are indicators of severe disease and generally require treatment. Other risk factors that should prompt treatment include immunosuppression, old age, pregnancy, diabetes, pre-existing cardiopulmonary conditions, and chemotherapy. Our patient was initially not treated due to immunocompetency and lack of extrapulmonary signs and symptoms, but once the patient presented with disseminated infection, fluconazole and AmB were administered per IDSA guidelines.4
Dermatologists should consider disseminated coccidioidomycosis infection in patients with solitary or few violaceous or verrucous papules, plaques, or nodules with sinus tracts, as well as travel to endemic regions.
References: 1. Arce M, Gutierrez-Mendoza D. Primary and disseminated cutaneous coccidioidomycosis: clinical aspects and diagnosis. Curr Fungal Infect Rep. 2016;10:132-139. 2. Garcia Garcia SC, Salas Alanis JC, Flores MG, Gonzalez Gonzalez SE, Vera Cabrera L, Ocampo Candiani J. Coccidioidomycosis and the skin: a comprehensive review. An Bras Dermatol. 2015;90(5):610-9. 3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-23. 4. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-46.






Case 35: Hyalohyphomycosis in an Immunocompromised Patient with Concurrent Prostate Cancer and Acute Myelogenous Leukemia
Caregivers: Garrett Vick, MD; Aimee Coscarart, MD; Alun Wang MD, PhD New Orleans, LA
History: A 74 year old Caucasian man diagnosed with prostate cancer in July 2018 and subsequent diagnosis of AML in August 2018 for which patient received cytarabine and anthracycline. Post-chemotherapy, he developed complications including neutropenic fever, sepsis, and fungemia with Trichosporon beigelli. The patient was treated with broad antimicrobials (vancomycin, meropenem, acyclovir, voriconazole, and amphotericin B). Dermatology was consulted for rash of red bumps to chest that had been present since admission (3 weeks) and prior to chemotherapy or other medications administered in hospital. Bumps were unresolved with antimicrobials. Skin lesions of the left chest were biopsied and sent for H&E as well as AFB/Bacterial/Fungal cultures.
Physical Examination: General appearance: chronically ill appearing, alert, awake, oriented, patient appeared ill. Skin: Multiple 2-3mm erythematous non-follicular based papules diffusely distributed to chest, back, abdomen, and extremities. No purpura, vesicles, pustules, or scale noted. PICC line to right upper extremity.
Laboratory Data: LDH 3032 UNITS/L (normal 87 - 241 UNITS/L); WBC (normal 4.5 - 11.0K): 268.2K; Smear: 1+ for Smudge Cells, Poikilocytosis, Anisocytosis, Macrocytosis, and Ovalocytes; Hgb (normal 13.5 - 17.5 GM/DL): 9.0 GM/DL; Hct (normal 41.0 - 54.0 %): 27.8% Plt (normal 160 – 420K): 40K
Histopathology: Skin, left chest, biopsy: Deep fungal infection concerning for hyalohyphomycosis; culture necessary for species identification.
Clinical Course: Antibiotics, antivirals, and antifungals were continued with plan to tailor therapy according to pathology and culture results. However, patient expired less than twenty-four hours following biopsy. Bacterial, fungal, and acid fast bacilli cultures remained negative with no organisms isolated.
Diagnosis: Hyalohyphomycosis
Points of Emphasis: Deep mycoses can be subcutaneous or systemic.1 Inoculation occurs via penetration of dermis by debris contaminated with organisms, or indirectly with spread of organisms to the skin from another portal of entry (eg. respiratory tract).1-2 Immunosuppression is a well cited predisposition for systemic involvement.1-6
Hyalohyphomycosis is a categorical term that does not refer to infection with a specific organism, but instead to an infection that is caused by non-pigmented, hyaline fungi, with incriminated genera including Acremonium, Fusarium, Purpureocillum, Scopulariopsis, Penicillium, Paecilomyces and Pseudallescheria.1-4,6 While immunosuppression is often associated with systemic spread, it is not required.1,4 The clinical presentation may include fever, pneumonia, pleuritic chest pain, necrotic plaques or ulcers.2 Given the non-specific nature of these symptoms and the fact that severely immunocompromised patients may harbor more than one infectious agent, the differential should also include cryptococcosis, histoplasmosis, and other systemic mycoses.1,7
The diagnosis of hyalohyphomycosis can be suggested by the presence of hyaline hyphae on microscopy, but confirmation of the diagnosis and identification of the causative organism to ensure proper treatment requires further investigation such as with subsequent culture (usually with Sabouraud’s dextrose agar without antibiotic or growth inhibitors).1-2,8 A negative fungal culture may not be reliable if it is not performed using the correct media or if the required growth parameters are not meticulously ensured. It is also important to not erroneously discount the growth of a rarely encountered organism as merely a contaminant.3 Ultimately, even if a culture is performed correctly, it is still possible that the organism may not be successfully grown.8 One option to further help identify organisms is by utilizing quantitative PCR of ribosomal DNA from formalin-fixed paraffin-embedded tissues which has been demonstrated as possible with Fusarium and Scedosporium with 64% sensitivity.8
Treatment options depend on the organism and corresponding susceptibility, patient characteristics and infection location.9-11 These include surgical excision, terbinafine, triazoles (itraconazole, voriconazole, posaconazole) and amphotericin B .2,11 For immunocompromised individuals, triazoles are often used in combination with amphotericin B for extended regimens (approximately 6 months).1-2,5,10 However, some organisms may be resistant to even combined use of amphotericin B and triazoles as seen in previous cases of disseminated Fusarium as well as described here in our case due to an unidentified fungal agent.5,9-11
Thanks to the advances in transplant medicine as well as other fields that employ immunosuppressive agents, the incidence of deep mycoses, including cases of hyalohyphomycosis has increased over the last few decades.10 Important factors to emphasize include that immunocompromised patients can have multiple conditions (more than one type of malignancy) and can be infected with multiple infectious agents simultaneously (Trichosporon beigelli, hyalohyphomycosis). Hyalohyphomycosis can have similar presentation amongst the different known causative species, requiring verification via methods beyond microscopic evaluation. Finally, it is also possible that once the correct organism is identified, it may demonstrate resistance to currently available antifungal regimens.
References: 1. Carrasco-Zuber JE, Navarrete-Dechent C, Bonifaz A, Fich F, Vial-Letelier V, Berroeta-Mauriziano D. Cutaneous Involvement in the Deep Mycoses: A Literature Review. Part I- Subcutaneous Mycoses. Actas Dermosifiliogr. 2016. Dec; 107(10):806-815. doi: 10.1016/j.ad.2016.05.017. 2. Perusquía-Ortiz AM, Vázquez-González D, Bonifaz A. Opportunistic filamentous mycoses: aspergillosis, mucormycosis, phaeohyphomycosis and hyalohyphomycosis. J Dtsch Dermatol Ges. 2012. Sep;10(9):611-621. doi: 10.1111/j.1610-0387.2012.07994.x. 3. Ajello, L. Hyalohyphomycosis and phaeohyphomycosis: two global disease entities of public health importance. Eur J Epidemiol. 1986. Dec;2(4):243-251 4. Das S, Saha, R, Dar SA. et al. Mycopathologia. (2010) 170: 361. https://doi-org.libproxy.tulane.edu/10.1007/s11046-010-9334-1






