2017 Clinical & Dermatopathologic Cases
- 15. Atypical (Symplastic) Piloleiomyoma with Follicular Induction
- 16. Dystrophic Calcinosis Cutis
- 17. Osteoma Cutis with Gelatinous Transformation
- 18. Pigmented Fungiform Papillae of the Tongue
- 19. Massive Localized Edema of the Vulva
- 20. Anaplastic Plasmacytoma
- 21. Angioinvasive Lymphomatoid Papulosis
- 22. Lesser known CD8+ Cutaneous T-Cell Lymphomas
- 23. Henoch Schonlein Purpura
- 24. Acute Generalized Exanthematous Pustulosis secondary to IVIG
- 25. Granulomatous Folliculitis as a Wolf's Isotopic Response
- 26. Acute Radiation Dermatitis with Koebnerized Psoriasis
- 27. Unmasking Lupus with Imiquimod Therapy
- 28. Palmarplantar Eruption on Nivolumab
- 29. Keratosis Lichenoides Chronica
- 30. Paraneoplastic Psoriasiform Necrolytic Erythema
- 31. Rowell's Syndrome
- 32. H Syndrome
- 33. Capillary Malformations with RASA1 Mutations
- 1a. Secondary Syphilis
- 1b. Granulomatous Syphilis
- 2. Furuncular Cutaneous Myiasis caused by Dermatopia Hominins
- 3. Norcardia Mycetoma
- 4. Cutaneous Cryptococcus neoformis
- 5. Subungual Amelanotic Malignant Melanoma
- 6. Sun's Nevus (Acquired Unilateral nevus of Ota)
- 7. Verrucous Carcinoma
- 8. Red Dot Basal Cell Carcinoma
- 9. Atypical Carcinoid Tumor of the Thymus, Metastatic to Skin
- 10a. Adult-Onset Langerhans Cell Histiocytosis
- 10b. Langerhans Cell Sarcoma
- 11. Clear Cell Sarcoma of the Large Toe
- 12. Angiosarcoma at the Site of a Previous Dialysis Graft
- 13. Pleomorphic Fibroma
- 14. Plexiform Neurilemmoma Arising in a Surgical Scar
Case 1a: Secondary Syphilis
Caregivers: Nicholas Guido, B.S., Lacey Sullivan, M.D., Virginia Alldredge, M.D. MPH, Katherine Brag, M.D., Alun Wang, M.D., Ph.D., New Orleans, LA
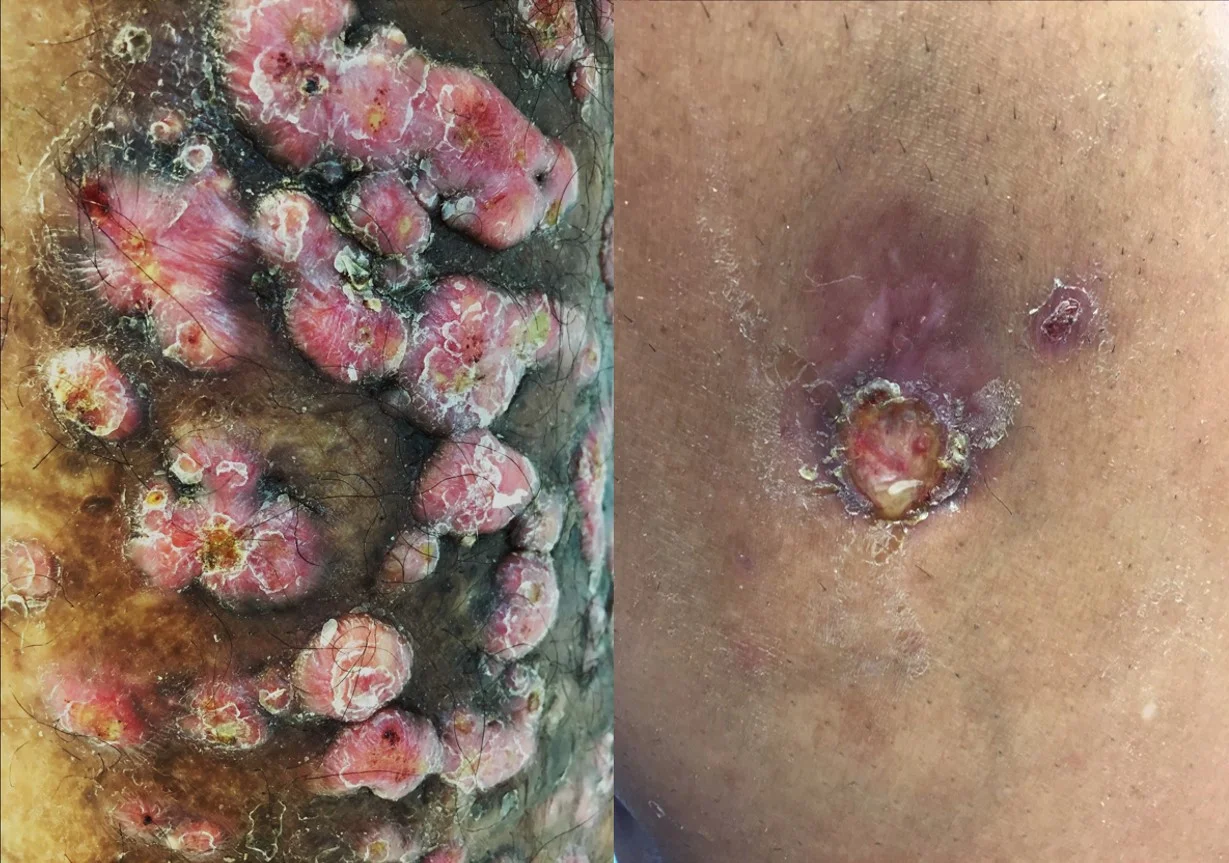
History: 25 year-old African American male presented to the Emergency Department with a rash of two weeks duration. The rash initially began on his scrotum and then spread to his arms and legs including his palms and soles. He had no oral lesions. Patient reports unprotected sex with new partners for the past six months including sex with men. Drug use history is significant for marijuana but no other illicit drugs. He denied having any other medical problems. The patient also reported an additional “fungal rash” on his upper chest and proximal arms which had been treated with pills and creams. This rash recurred annually and had been stable.
Physical Examination: Firm, smooth, erythematous to violaceous flat papules on the scrotum, shaft of penis, left lower extremity, right lower extremity, trunk, palms and soles. Hypopigmented, well demarcated plaques with inducible scaling on the back, abdomen, right upper extremity and right lower extremity.
Laboratory Data: ALT -214; AST -136; Alkaline phosphatase -379; ELISA for HIV- negative; RPR pending at time of dermatology consult.
Histopathology: Skin with a neutrophilic infiltrate in the upper dermis accompanied by vascular dilation and dermal edema. Overlying epidermis with mild spongiosis and parakeratosis containing neutrophils. Focal neutrophilic pustules within the epidermis. An immunohistochemical study for treponema pallidum showed numerous spirochete organisms within the epidermis and at the dermal epidermal junction with scattered organisms in the papillary dermis. A PAS stain was negative for fungal elements.
Clinical Course: Patient was treated empirically with Penicillin 2.4 million units intramuscularly, and planned to follow up 2 weeks later for additional dose. RPR was found to be reactive following patient ED discharge. Case was reported to appropriate authorities.
Diagnosis: Secondary Syphilis
Points of Emphasis: Syphilis is a systemic sexually transmitted disease caused by Treponema pallidum. It has diverse clinical manifestations depending on stage of infection and the organ system(s) involved. Coinfection with other sexually transmitted diseases is common, therefore appropriate testing is also recommended. Primary syphilis manifests as an ulcer or chancre at the infection site, however this may be asymptomatic and undetected. Secondary syphilis has many systemic symptoms—rash, mucocutaneous lesions, and adenopathy are most common. Latent syphilis is asymptomatic, but may be detected by serologic tests; it is classified as early (if acquired within the previous year), late, or of unknown duration. Tertiary syphilis may manifest with neurologic or less commonly with gummatous or cardiovascular symptoms. Secondary syphilis occurs weeks to months after primary syphilis and manifests as multisystem involvement due to bacteremia. Usually presents with a generalized rash (never vesicular, except in congenital syphilis) that is macular, papular, or pustular, or a combination of all three; mucocutaneous lesions, generalized lymphadenopathy, and nonspecific systemic symptoms are common. Central nervous system involvement is found in up to 40% of patients at this stage. The disease is more common in men than women. Secondary syphilis should always be suspected in patients with an undiagnosed rash or mucosal eruption, especially if accompanied by generalized lymphadenopathy or if the patient is at risk for syphilis. Nontreponemal and treponemal tests are almost always positive during this time. False negative RPR/VDRL may occur due to the prozone phenomenon; retesting after dilution of the sample may yield a positive result. Diagnosis can be confirmed by direct visualization of T pallidum, although the vast majority of cases at this stage are diagnosed by serology and physical examination alone. The USPSTF recommends screening for syphilis in all nonpregnant adults and adolescents at increased risk for infection (Grade A recommendation).
References: 1. Abell, E., R. Marks, and E.W. Jones, Secondary syphilis: a clinico-pathological review. Br J Dermatol, 1975. 93(1): p. 53-61. 2. Bolognia J, Jorizzo JL, Schaffer JV.Dermatology.3rd ed. London; Philadelphia: Elsevier Saunders; 2012. 3. US Preventive Services Task Force (USPSTF). Screening for Syphilis Infection in Nonpregnant Adults and Adolescent:US Preventive Taskforce Recommendation Statement. JAMA. 2016:315(21):2321-2327.






Case 1b: Granulomatous Syphilis
Caregivers: Danielle de Stefano, M.D., Melissa Dillard, M.D. Juany Garza, M.D., Joseph Susa, D.O., Clay Cockerell, M.D., Dallas, TX
History: 25 year-old African-American female presented with a diffuse rash which developed over the course of eight weeks. She reported that the rash had begun on her extremities, rapidly spread to the face and trunk, and was extremely itchy and painful. Prior to rash onset, she experienced flu-like symptoms, which included fever, arthralgia, myalgia, and sore throat. In addition, at the time of initial visit, the patient had shortness of breath, chest pain, and joint and muscle pain. She had been seen in three different medical facilities and treated with NSAIDS, antihistamines, and hydrocortisone cream with no improvement. Upon further questioning, she reported having a new sexual partner approximately 6 weeks prior to symptom onset. Two weeks following this sexual encounter, she developed dysuria and a genital “sore.” Her past medical history also included gonorrhea and trichomoniasis.
Physical Examination: Multiple, disseminated erythematous, and indurated papules (4-8 mm) on the face, neck, trunk and extremities. Discrete lesions were present on the palms and soles of the feet, with a noticeable collarette of scale. Genital examination revealed two superficial ulcers on the labia minora measuring 4-10 mm in diameter. Inguinal lymphadenopathy was also seen.
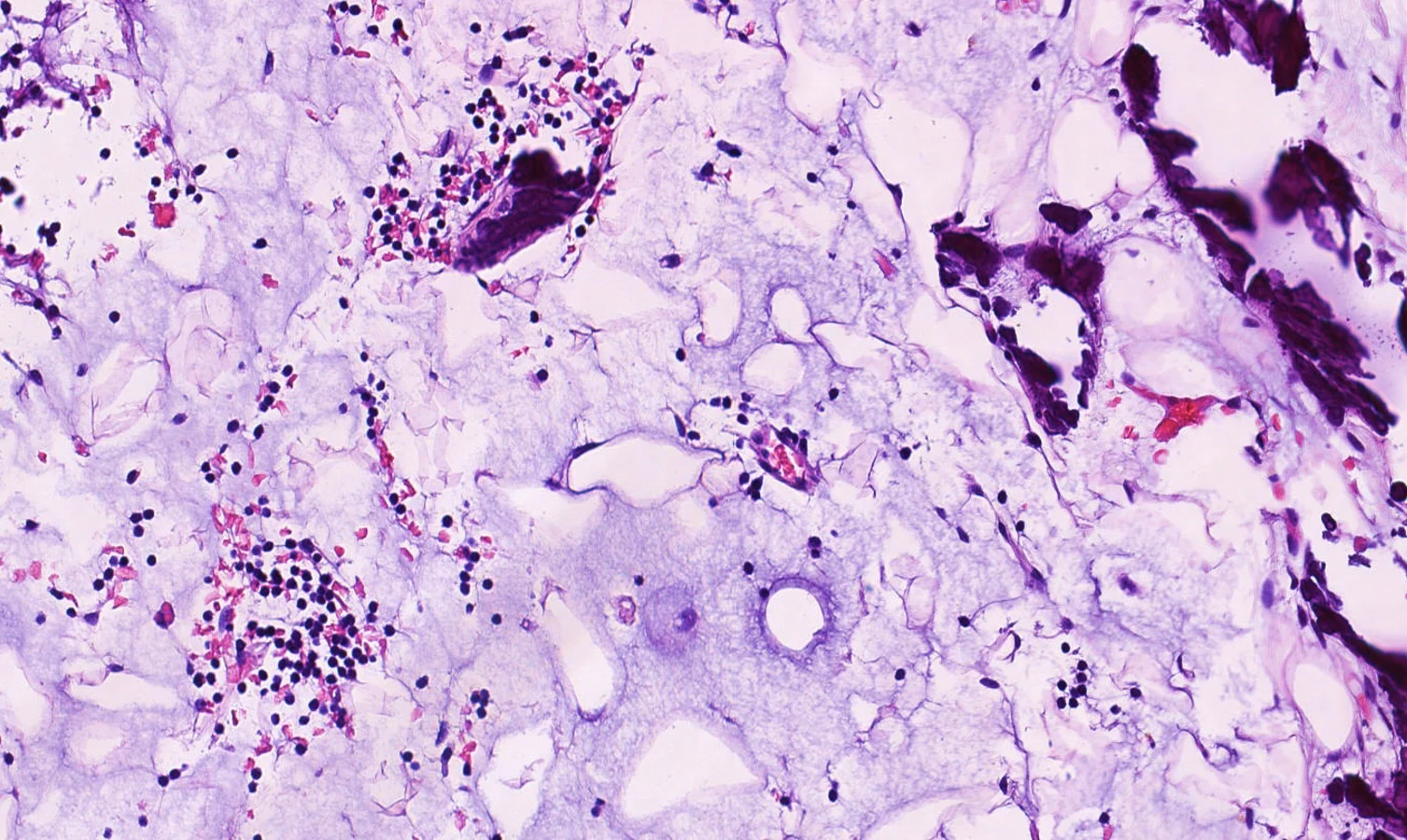
Histopathology: Superficial and deep inflammatory infiltrate consisting of lymphocytes, histiocytes, and plasma cells. Suppurative granulomatous inflammation was also seen, with scattered eosinophils and multinucleated giant cells. The infiltrate was predominantly perivascular and periadnexal and the epidermis was thinned with some areas of spongiosis and overlying crust. Special stains for PAS, FITE and Gram were negative. An immunohistochemical stain for T. pallidum confirmed the presence of spirochetes.
Clinical Course: The treatment consisted of oral doxycycline 100 mg twice a day for 14 days; Naproxen 500 mg twice a day for 5 days; Clobetasol 0.05% twice a day on affected areas; and follow-up consultation in 3 weeks. She was also screened her for chlamydia, gonorrhea, herpes and HIV and the tests were negative. On her follow-up appointment, after 14 days of treatment with good compliance, she demonstrated great response with clearing of all lesions and improvement in her joint pain. Post-inflammatory hyperpigmentation was seen.
Diagnosis: Granulomatous syphilis
Point of Emphasis: Granulomatous syphilis is one of the less common but exuberant presentations of secondary syphilis. The histopathological appearance of secondary syphilis can be as varied as and just as challenging as its clinical presentation. It is considered one of the great “histopathological imitators”. The most common histologic presentation consists of a plasma cell-rich infiltrate with endothelial proliferation and swelling. However, psoriasiform, spongiotic, lichenoid, and perivascular patterns have also been described. No single histological finding is pathognomonic for this disease. Therefore, a high level of suspicion is essential whenever a dermal-based granulomatous infiltration is identified, especially in the setting of an increased number of plasma cells, even if slight. Mixed and multiple patterns of inflammation should also raise suspicion. Further studies, preferably immunohistochemistry for Treponema pallidum, should then be performed in order to exclude this entity.
References: 1. Rysgaard C, Alexander E, Swick B. Nodular secondary syphilis with associated granulomatous inflammation: case report and literature review. J Cutan Pathol. April 2014;41(4):370-379. 2. Liu, J. and Ma, D. (2014), Disseminated nodular and granulomatous secondary syphilis. J Dermatol, 41: 650–651.







Case 2: Furuncular cutaneous myiasis caused by dermatobia hominins
Caregivers: Skylar Souyoul, M.D., Pam Martin, M.D., Elizabeth Bucher, M.D., Stephen Lambert, M.S.-IV, New Orleans, LA
History: A 35-year-old male presented to his general dermatologist for two, painful, growing nodules on his scalp. The nodules had been present for one month. The patient denied any known bites or systemic symptoms. Travel history included a trip to Belize two months prior, which involved camping in the jungle.
Physical Examination: Two discrete, tender, semi-firm subcutaneous nodules, each with overlying punctum on the left occipital scalp.
Histopathology: Suppurative and granulomatous dermatitis, probably secondary to a ruptured follicular cyst. Deep dermal and subcutis inflammatory cell infiltrate of neutrophils, histiocytes, and lymphocytes accompanied by edema. No follicular wall was identified.
Clinical Course: The patient was referred to a Mohs surgeon for excision of suspected follicular cysts. An elliptical incision was made in the skin over one of the subcutaneous nodules to include the punctum. Dissection around the nodule revealed an insect larva. The larva and overlying excised skin were submitted for histopathology. New pathology showed “Inflamed granulation tissue with pseudocyst, foreign body granulomatous reaction and arthropod larva.”
Diagnosis: Furuncular cutaneous myiasis caused by Dermatobia hominis, the human botfly.
Points of Emphasis: Furuncular myiasis caused by Dermatobia hominis, the human botfly, is endemic throughout Central and South America. The lesions often resemble a furuncle and are commonly misdiagnosed as a bacterial skin infection. Female D. Hominis lay their mature eggs on blood-feeding arthropods such as flies or mosquitoes. These eggs then hatch when the fly or mosquito lands on a warm-blooded host. The hatched larvae then enter the skin through the insect bite or via hair follicles. The larvae matures in the subdermal cavity for 4-18 weeks and can grow up to 2 cm in length. Furuncular myiasis lesions are usually located on exposed skin such as the limbs, head, and neck. Initially, the lesion resembles a pruritic, painful, mosquito bite, that later develops into a small erythematous nodule with a central pore which can exude serosanguinous discharge. The central punctum allows the larva to breathe while it burrows deeper into the skin forming a dome-shaped nodule. The lesions are not responsive to antibiotic therapy and are commonly misdiagnosed as folliculitis, insect bites, or impetigo. Traditional treatments for D. Hominis myiasis involve the removal of the larva by attempting to asphyxiate it occluding its breathing opening with wax, paraffin, nail polish, or sealing ointment for 24 hours, which forces the larva to burrow upwards to allow for easy extraction with forceps. This conservative method is only possible during the early stages of development because as the larva matures its concentric spines anchor it firmly into the tissue. If conservative methods fail, the best management involves making a small incision through the opening and pulling the larva out with a hemostat. After the larva extraction, the wound should be cleaned and debrided along with tetanus immunization. If the larva dies and remains within the subdermal tissue, a foreign body granuloma or secondary infection can occur. Antibiotics are not usually recommended if no secondary infection exists.
References: 1. Bhandari R, Janos DP, Sinnis P. Furuncular myiasis caused by Dermatobia hominis in a returning traveler. Am J Trop Med Hyg. 2007;76:598–599. 2. Maier H, Honigsmann H. Furuncular myiasis caused by Dermatobia hominis, the human botfly. J Am Acad Dermatol. 2004;50(2 Suppl):S26-S30. 3. Robbins K, Khachemoune A. Cutaneous myiasis: a review of the common types of myiasis. Int J Dermatol 2010;49; 1092-8.





Case 3: Norcardia mycetoma
Caregivers: Hailey Rouhana M.D., Gaston DelaBretonne M.D., New Orleans, LA
History: 40-year-old Honduran man with no significant past medical history presented with lesions on his right thigh for 7 years. He reported some of the lesions resolved, but new ones developed. The lesions progressively worsened in the past 6 months. Associated symptoms included mild tenderness and pruritus. He reported fevers for one week and an 18 pounds weight loss over the past year. He denied known trauma to the area, exposure to animals, or contacts with similar lesions. He works as a mechanic and lived in California for the past 15 years. He previously lived in Honduras and travels back once a year.
Physical Examination: Examination revealed a well-demarcated area of the right lateral and posterior thigh with scaly erythematous fluctuant plaques and papules containing small pustules and some areas of overlying yellow crust. Surrounding hyperpigmentation was noted. The medial right thigh had 2 ulcerated sinus tracts with translucent yellow exudate. The right thigh was significantly larger in diameter than the left thigh. Right inguinal lymphadenopathy was appreciated.
Laboratory Data: CBC revealed leukocytosis of 14,700 with 74% neutrophils; low hemoglobin and hematocrit of 9.1 and 28.6 respectively; high platelets of 430. CMP was unremarkable. CRP and ESR were elevated at 14.9 and 104 respectively. HIV testing was negative. Bacterial tissue culture revealed light growth of methicillin-sensitive Staph aureus. Tissue cultures for fungal and acid-fast organisms were negative. Blood and urine cultures were negative. Cryptococcal antigen, Histoplasma antigen, Blastomyces dermatitidis antigen, T-spot, and complement fixation for Coccoides immitis were negative. Ultrasound revealed extrinsic compression of the right femoral vein and an ill-defined complex mass disrupting the normal muscular and soft tissue planes in the anterior and medial thigh. Two large lymph nodes were noted in the right femoral region. MRI of the right lower extremity revealed a large ill-defined mass-like structure involving the entire thigh, tracking along the fascial planes, and partially replacing the vastus lateralis, the gluteus maximus, and the muscles of the adductor compartment. Numerous areas of extensive subcutaneous and cutaneous fistularization/tract formation with sites of cutaneous eruption were noted. No features of osteomyelitits were seen.
Histopathology: Organizing abscess with focus of gram positive, Fite positive, and GMS positive filamentous bacteria (compatible with Nocardia); granulation tissue, granulomatous and lipophagic granulomatous inflammation, and marked fibrosis.
Clinical Course: Patient is currently being treated with Bactrim with significant improvement of mycetoma.
Diagnosis: Nocardia mycetoma.
Points of Emphasis: Bacterial (actinomycotic) mycetomas are rare tumor-like infections of the skin and subcutaneous tissue. Nocardia are the most common cause and are ubiquitous, gram-positive, modified acid-fast, filamentous branching bacteria. Typically, penetrating trauma and subsequent contamination with soil or vegetation leads to infection. Mycetomas begin as a papule or nodule and slowly expand causing local destruction and eventually fistulae with intermittent purulent exudate often containing granules. They usually occur on the feet and lower extremities where trauma is more likely to occur. The majority of mycetomas are asymptomatic. Constitutional symptoms and systemic spread are uncommon. The process evolves slowly and may progress over decades. The diagnosis of Nocardia can be made by culture with routine media but may require longer incubation time. Histopathology may also lead to the diagnosis as it did in this case. MRI is useful for establishing the extent of the lesion and has greater sensitivity than radiographs, ultrasound, or CT. Treatment of Nocardia is Bactrim DS twice a day for a minimum of 6 months. If Bactrim is contraindicated, minocycline can be used.
References: 1. Zijlstra EE, van de Sande WW, Welsh O, Mahgoub el S, Goodfellow M, Fahal AH. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016 Jan;16(1):100-12. 2. Chen B, Tang J, Lu Z, Wang N, Gao X, Wang F. Primary Cutaneous Nocardiosis in a Patient With Nephrotic Syndrome: A Case Report and Review of the Literature. Medicine (Baltimore). 2016 Jan;95(3):e2490.



Case 4: Cutaneous Cryptococcus neoformis
Caregivers: Aubrey Allen B.A., Jaclyn N. Hess, M.D., Daniel J. Sheehan, M.D., Loretta S. Davis, M.D., Augusta, GA
History: A 50-year-old transgender female with HIV, non-compliant with medications, presented with headache and nausea of two weeks duration. Prior to the headache she had a solitary “bump” on the back of her neck. Numerous “bumps” subsequently came up, predominantly on the anterior neck, face and a few on the bilateral hands and legs. She denied travel or exposure to birds or bats.
Physical Examination: Face, neck and upper chest had scattered 2-4 mm umbilicated papules with central crusting. A few solitary papules were present on bilateral hands and lower legs.
Laboratory Data: HIV viral load was elevated at 243,567 RNA copies/ml. CSF india ink was stained positive for encapsulated yeast forms. Cryptococcal antigen was positive at a titer of 1:2560. Eventually Cryptococcus grew from tissue culture and blood culture.
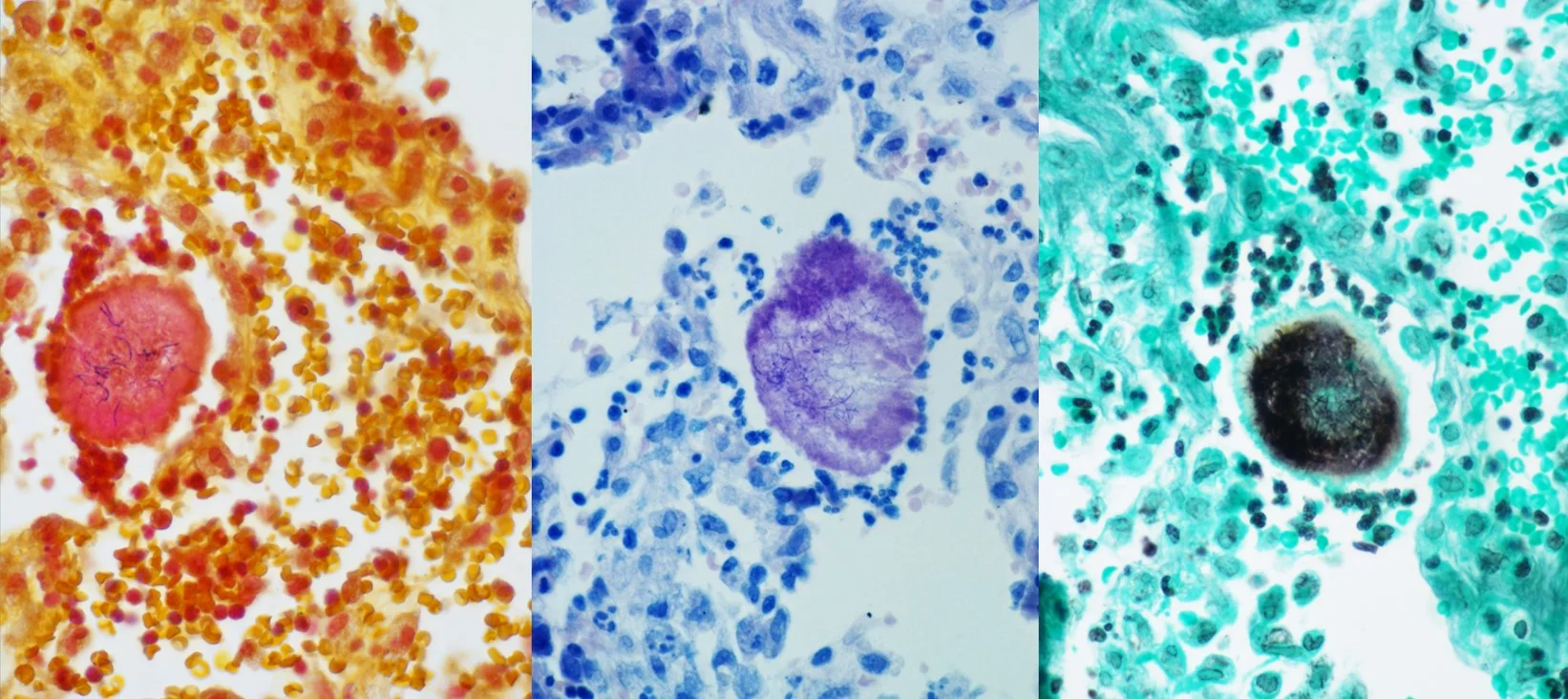
Histopathology: Numerous pleomorphic yeast were seen clustered within clear spaces. GMS: positively stained numerous yeast-like fungal structures throughout the epidermis and dermis. PAS-FG: positively stained numerous yeast-like fungal structures throughout the epidermis and dermis. Mucicarmine: positively stained the yeast-like structures.
Clinical Course: The patient received IV amphotericin and oral flucytosine. He signed himself out of the hospital against medical advice 10 days later with a prescription for fluconazole.
Diagnosis: Cutaneous Cryptococcus neoformans
Points of Emphasis: Cryptococcus neoformans is an encapsulated, yeast-like fungus found in dried avian and bat excreta. When inhaled, the organism causes the formation of pulmonary granulomas and pneumonia-like illness and can also present clinically as meningoencephalitis. Infection usually occurs in immunocompromised individuals, especially AIDS or organ transplant recipients. Skin lesions can be the first presentation of a systemic infection. Cryptococcus can present with a variety of skin manifestations such as acneiform lesions, molluscum-like lesions, purpura, vesicles, nodules, abscesses, ulcers, granulomas, pustules, cellulitis and subcutaneous abscesses. Most often, cutaneous Cryptococcus presents secondary to disseminated disease, and is present in 10-15% of patients with systemic cryptococcosis. Disseminated disease, in patients with HIV most commonly presents cutaneously as umbilicated lesions resembling Molluscum contagiosum on both clothed and exposed areas, particularly the head, neck and forearms. Diagnosis may be made by examination of India ink preparations of aspirates or by Tzanck smear. The organisms may be isolated on Sabouraud’s agar and are mucicarmine positive, doubly refractile and do not stain with Congo red. Alcian blue-PAS stain contrasts the cell wall and capsule. Histologically, Cryptococcus presentation is variable, ranging from tuberculoid granulomas in the dermis and subdermis with few organisms, to an extensive mucoid mass formed by many organisms surrounded by their mucinous capsules. A common pattern is dense inflammatory cell infiltrates with multinucleate giant cells containing several organisms with refractile walls.
References: 1. Srivastava, G. N., Tilak, R., Yadav, J., & Bansal, M. (2015). Cutaneous cryptococcus: Marker for disseminated infection. BMJ Case Reports, 2015. 2. Patterson, James W. 1946- (James Willis), & Hosler, G. A. (2016;2014;). Weedon's skin pathology (Fourth ed.). Philadelphia, PA: Churchill Livingstone/Elsevier.





Case 5: Subungual Amelanotic Malignant Melanoma
Caregivers: Danielle de Stefano, M.D., Melissa Dillard, M.D., Elena Maxim, MS-IV, Yevgeniya Byekova Rainwater, M.D., Joseph Susa, D.O., Clay Cockerell, M.D., Dallas, TX
History: A 42-year-old woman with a history of right thumbnail dystrophy for six years and remote history of trauma presented to the clinic. Multiple nail clippings had been submitted for analysis in the past, however, all had come back negative for hyphae. Despite empiric treatment for fungal infection over the past one year, there was no significant improvement in her nail findings. During her initial visit with us, once more a nail clipping was performed. Tavaborole was subsequently prescribed, and the patient was instructed to perform weekly white vinegar soaks. At her six-month follow up appointment, she reported some improvement in nail appearance. She was instructed to continue treatment and return for follow up in six months’ time. Before her scheduled appointment, the patient called to report that her “nail fell off”.
Physical Examination: On her initial visit, distal onycholysis with prominent subungual debris at the right first digit was seen. All other fingernails were normal in appearance. On examination during her 3rd visit (after nail loss) a small, skin-colored, subungual papule with focal ulceration was noted.
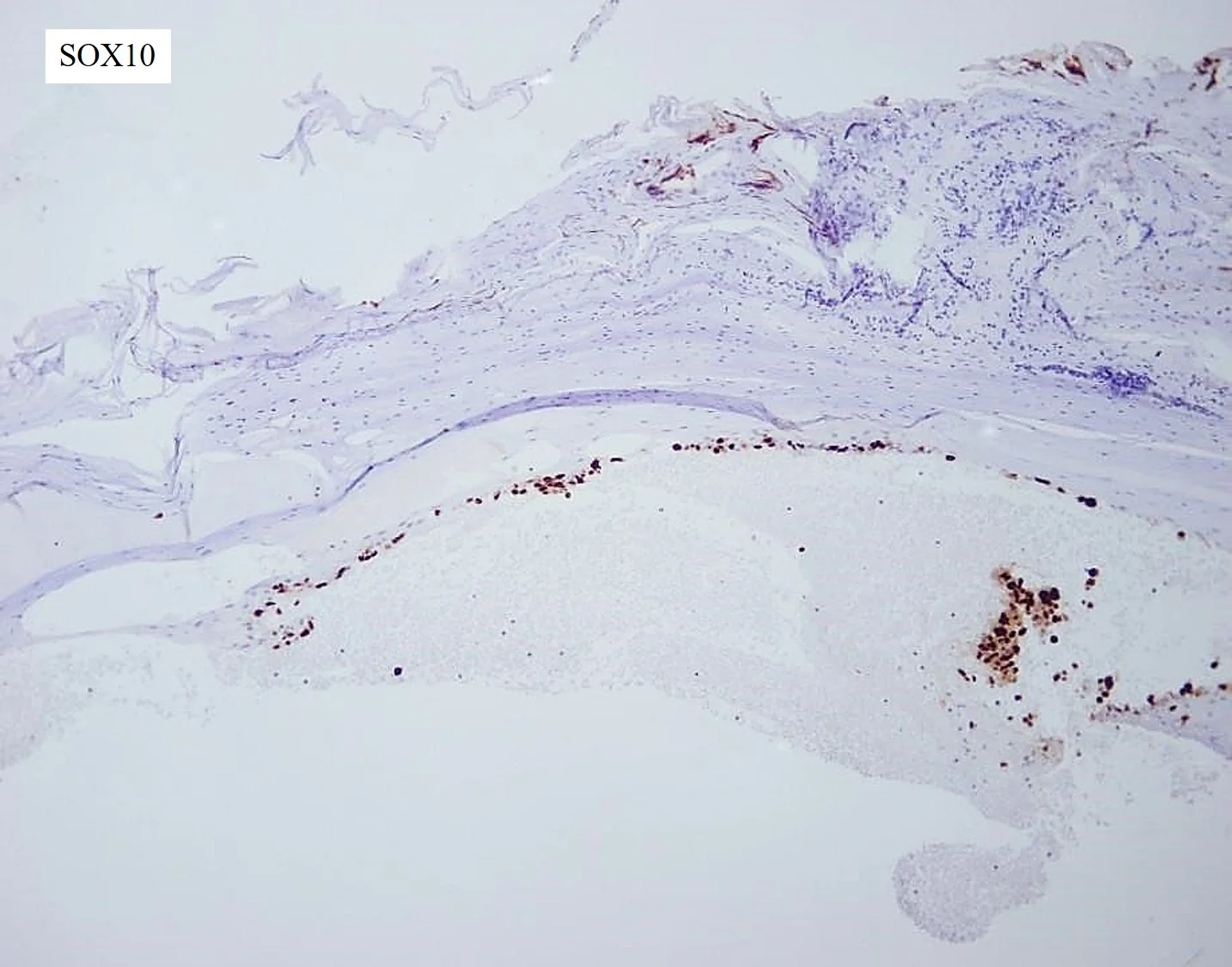
Histopathology: The histologic examination of the nail clipping and culture did not show significant abnormalities. The subsequent shave biopsy demonstrated edema of the corneal layer with neutrophilic aggregates and an underlying traumatic hemorrhage. Dyscohesive, atypical cells with hyperchromatic nuclei and scant cytoplasm arranged in an interrupted ribbon pattern were noted at the dermal-epidermal junction (DEJ). While most of the cells appeared confined to the epithelium, there were a few, atypical loose cells underneath the DEJ. Immunohistochemical stains for SOX-10 were strongly positive, confirming the diagnosis of malignant melanoma of the nail apparatus. The lesion was 0.3mm in thickness. The Mohs specimen revealed a large and asymmetric infiltrating tumor with melanocytes present in large solid sheets and nodular, shapeless masses extending diffusely through subepithelial connective tissue.
Clinical Course: Treatment included finger amputation distal to the PIP joint and lymph node dissection, which was negative. She had a negative PET scan and continued to be monitored by dermatology, surgical and medical oncology.
Diagnosis: Subungual amelanotic malignant melanoma, at least 3.5mm in thickness, Clark level IV; all margins were positive with a mitotic index of 2/mm2. Perineural involvement was also noted.
Points of Emphasis: Amelanotic, subungual melanoma (SUM) is often misdiagnosed and untreated for prolonged periods of time due to its resemblance to other benign entities. Misdiagnosis rates up to 85% have been reported in non-dermatologists. This was the case with our patient, who had an untreated subungual melanoma for over seven years. The initial changes in subungual melanoma involve pigmentation of the nail bed, however this finding is only observed in 76% of cases. Approximately one third of cases are detected during this phase. Secondarily, nail bed changes often accompanied by secondary infection or ulceration with granulation tissue in the nail bed are observed. At diagnosis the tumor is often advanced, and the majority of cases are usually detected at Clark levels IV or V. Due to the delay in diagnosis, survival rates for SUM patients are considerably worse than other melanoma forms. Prognosis in SUM is determined by tumor thickness and five-year survival is estimated at 10% to 30%. The differential diagnosis for SUM includes multiple malignant and benign entities. Benign nail plate lesions such as verruca vulgaris, traumatic onychodystrophy, pyogenic granuloma, Glomus tumor, onychopapilloma and subungual keratoacanthoma can resemble SUM. Squamous cell carcinoma and basal cell carcinoma are also differential diagnoses for skin colored nodular lesions on the nail plate. Suspicious diagnostic findings such as nail fold pigmentation (Hutchinson’s sign), lifting off of the nail from the nail bed, and non-healing ulcerating lesions should raise clinical suspicion for melanoma. Adequate biopsy specimens, which may be technically difficult to obtain, are necessary for correctly diagnosing SUM. If high clinical suspicion for melanoma exists, a full thickness excisional biopsy is recommended, as well as nail removal. Despite the absence of clinical pigmentation in amelanotic tumors of the nail, they may still demonstrate the presence of pigment on biopsy if special stains are used, suggesting that the biologic capacity for pigment production is present.
References: 1. Boyer A. Fungus hematode de petit doights. Gazette médicale deParis 1834;2:212. 2. Hutchinson J. Melanosis often not black: melanotic whitlow. Br Med J. 1886;1:491–494. 3. Arican O, Sasmaz S, Coban YK , Ciralik H. Subungual amelanotic malignant melanoma. Saudi Med J 2006;27:247-9.






Case 6: Sun's nevus (Acquired Unilateral Nevus of Ota)
Caregivers: Koriann Reed M.D., Mario Mitkov M.D., Mark Cappel M.D.; Jacksonville, FL
History: A 57-year-old man was evaluated for a pigmented lesion on his left temple. The lesion had been present for more than 5 years and was asymptomatic. The patient had not noted any recent changes in the size or color. He also denied previous external trauma or exposures to the site.
Physical Examination: Involving the left temple was a single, somewhat ill-defined, blue-grey hyperpigmented patch.
Histopathology: A punch biopsy of the left temple showed superficial dermal dendritic melanocytosis, consistent with nevus of Ota. Melan-A stain highlighted numerous dermal dendritic melanocytes oriented in a horizontal pattern. Fontana-Masson staining confirmed the melanin content within these dendritic melanocytes.
Clinical Course: Once the lesion was diagnosed, treatment options were reviewed with the patient but no further treatment was pursued.
Diagnosis: Sun’s nevus (acquired unilateral nevus of Ota)
Points of Emphasis: Nevus of Ota is a form of dermal melanocytosis that follows the distribution of the first two branches of the trigeminal nerve and is often present at birth or appears by adolescence. An acquired bilateral form of nevus of Ota-like patches has been described as Hori’s nevus and frequently involves middle-aged women of Asian descent. A case of an acquired unilateral nevus of Ota-like patch, also known as Sun’s nevus, was first reported in 1991. The histopathological features of an acquired dermal melanocytosis is identical to those of a congenital dermal melanocytosis, thus it is the clinical history that distinguishes the two entities. Q-switched lasers are a well-known treatment for nevus of Ota. A recent meta-analysis comparing the Q-switched alexandrite laser and the Q-switched Nd:YAG laser showed that the Q-switched alexandrite laser had both a higher treatment success rate and a lower complication rate.
References: 1. Whitemore S, et al. Late-onset nevus of Ota. Cutis 1991; 48: 213. 2. Mataix J, et al. Late-onset Ito's nevus: an uncommon acquired dermal melanocytosis. J Cutan Pathol. 2007 Aug;34(8):640-3. 3. Yu P, et al. Comparison of clinical efficacy and complications between Q-switched alexandrite laser and Q-switched Nd:YAG laser on nevus of Ota: a systematic review and meta-analysis. Lasers Med Sci. 2016 Apr;31(3):581-91.





Case 7: Verrucous Carcinoma
Caregivers: Carole Bitar, M.D., Kathryn Olivier, M.S., Julie Martin, M.D., Bethany Vincent, M.D., New Orleans, LA
History: A 41-year-old African-American male presented with a large, fungating mass on his left neck. The mass had been present since 1998. It began as the “size of a quarter” and had rapidly grown over the last six years, becoming painful. The patient also reported bleeding and purulent, foul-smelling drainage from the mass. He tried to tie a string around the base of the mass and experienced syncope. The mass felt “heavy” and also caused decreased range of motion and pain when he attempted to abduct the left shoulder. The patient denied fever, chills, weight loss, current medications, allergies or previous surgical procedures.
Physical Examination: 14 x 7 x 4 cm large, mobile, exophytic, polypoid, verrucous tumor on his left neck that was attached to the skin in a linear fashion along an 8 x 1 cm base. There was necrosis at the surface with purulent exudate. No palpable cervical lymphadenopathy was appreciated.
Laboratory Data: Skin culture was positive for Staphylococcus aureus and Proteus mirabilis for which the patient was given Bactrim BID x 10 days.
Histopathology: Verrucous tumor with both endophytic and exophytic components exhibiting papillomatosis, hyperkeratosis, parakeratosis and focal hypergranulosis. The rete ridges had a bulbous appearance and were composed of large, well-differentiated squamous epithelial cells. The acanthotic downgrowths extended focally to the middle reticular dermis and consisted of blunted projections with low mitotic activity. The fibrous stroma surrounding the epithelial downgrowths contained ectatic vessels and a mixed inflammatory infiltrate consisting of lymphocytes, histiocytes, neutrophils, plasma cells, and eosinophils.
Clinical Course: The patient was referred to surgical oncology for wide local excision under general anesthesia. The mass was completely excised. The wound defect measured 10 x 3 cm with primary closure.
Diagnosis: Verrucous carcinoma with negative margins.
Points of Emphasis: Verrucous carcinoma (VC) can be considered an uncommon variant of well-differentiated squamous cell carcinoma (SCC). VC most often presents in the oral cavity, genital area, or plantar surface and rarely is detected in the head and neck. When cutaneous VC is diagnosed, it is seen mostly in the 6th to 7th decades in patients with chronic inflammation, chronic infection, or scarring. While there is believed to be an association between Human Papillomavirus (HPV) infection and VC of the oral, ano-urogenital, and palmoplantar regions, studies that have investigated the association between cutaneous VC and HPV infection have yielded inconsistent results. In addition, the presence or absence of concurrent HPV has not yet affected the therapeutic strategies for treatment of VC. Clinically, VC presents as a verrucous hyperkeratotic plaque or nodule that may form sinus tracts and even erode into bone. The cutaneous variant of VC can be locally aggressive and is potentially associated with metastasis from cancers of the lymph nodes or skin. Treatment involves surgical excision with recommended 4mm clear margins. Other lesions similar to VC include papillary SCC, fibroepithelial polyp, seborrheic keratosis, verruca vulgaris, and pseudocarcinomatous hyperplasia. Histologically, VC is characterized by hyperkeratosis and/or hyperparakeratosis with minimal cell atypia and mitotic activity. There are lobules of well-differentiated squamous cells that arise from the epidermis and push deeply into the dermis and subcutaneous fat in a non-infiltrative manner. It enlarges in an expansive manner to form a well-circumscribed margin surrounding tissue that can appear as a multinodular, semi-pedunculated exophytic protrusion macroscopically. The nucleus may stain positive for Ki-67 and P53 protein in the proliferating basal layer, but the lack of staining does not it rule out. There is very limited data regarding additional work up in suspected patients. Because it is considered a variant of SCC, other imaging modalities, such as CT or MRI, used in advanced cutaneous SCC may also be beneficial in defining the extent of the disease. CT is used to determine the presence of invasion or lymph node metastasis, while MRI can assess perineural invasion.
References: 1. Kurisu, Yoshitaka, et al. “Immunohistochemical findings and differential diagnosis of papillary-type cutaneous verrucous carcinoma of the neck: A case report.” Oncology Letters, 20 May 2015, pp. 3823-3825. 2. Brinster, Noonshin K. Dermatopathology. Philadelphia, PA, Elsevier Saunders, 2011. 3. Costache, Mariana, et al. “Cutaneous verrucous carcinoma – report of three cases with review of literature.” Romanian Journal of Morphology & Embryology, vol. 55, no. 2, 6 June 2014, pp. 383-388.





Case 8: Red Dot Basal Cell Carcinoma
CaregiversR: Philip R. Cohen, M.D., San Diego, CA
History: A 71-year-old man with a history of actinic keratoses, basal cell carcinoma and squamous cell carcinoma presented with a new lesion on his nose.
Physical Examination: A 2x2 mm flesh-colored papule with a central 1 x 1 mm red dot was observed on his left nostril. The lesion did not blanch when a glass microscope slide was pressed against it.
Histopathology: Nodular aggregates of basaloid tumor cells were seen extendeding from the overlying epidermis into the underlying dermis. Dilated vessels were present in the areas of solar elastosis in the papillary dermis adjacent to the tumor. Hair follicles and perifollicular lymphocytic inflammation was also present.
Clinical Course: The tumor was excised using the Mohs technique.
Diagnosis: Red dot basal cell carcinoma (BCC).
Points of Emphasis: Red dot BCC is a distinct variant of BCC that clinically can mimic a hemangioma or telangiectasia. To date, the characteristics of red dot BCC have been reported in 7 Caucasian patients (median age: 71 years), including 3 women, 2 men and 2 patients for whom the gender was not provided. All of the patients had a history of actinic keratoses and more than one non-melanoma skin cancer; 1 woman also had history of 2 melanomas. The nose was the most common site of red dot BCC; other sites were the back and the thigh. The tumors were < 1 x 1 cm (median: 3 x 3 mm). They appeared as a solitary, small, red macule or papule; some were surrounded by erythema or a flesh-colored papule. Diascopic evaluation, which typically results in blanching of a vascular lesion, can be misleading. Some of the red dot BCCs blanched after a glass slide was pressed against the tumor. The pathology of red dot BCC includes not only less aggressive BCC histology (nodular or superficial or both) but also tumors with an aggressive pattern of invasive tumor cell strands. The treatment of choice for red dot BCC is excision using the Mohs surgical technique. In some of the patients, substantial lateral spread of tumor was noted beyond the observed clinical margins. There has been no recurrence of red dot BCC following treatment. In conclusion, the appearance of a new red dot on a sun-exposed site in a patient with a personal history of actinic keratoses or non-melanoma skin cancer should prompt the clinician to not only consider the possibility of red dot BCC but also to perform additional evaluation of the skin lesion.
References: 1. Cohen PR. Red dot basal cell carcinoma: report of cases and review of this unique presentation of basal cell carcinoma. Cureus 2017;9(3): e1110. 2. Loh TY, Cohen PR. Red dot basal cell carcinoma: an unusual variant of a common malignancy. J Drug Dermatol 2016;15:645-647. 3. Cohen PR. Red dot basal cell carcinoma. J Clin Aesthet Dermatol 2017, in press.






Case 9: Atypical Carcinoid Tumor of the Thymus, Metastatic to skin
Caregivers: Terry Henges, PA-C, Elena Maxim M.S.-IV, Danielle de Stefano M.D., Melissa Dillard M.D., Charles Burke M.D., Clay Cockerell M.D.; Dallas, TX
History: A 36-year-old female, with history of asthma and depression, initially presented to her primary provider with a mediastinal mass. A biopsy of the mass was consistent with an atypical, thymic, carcinoid tumor. Subsequent CT and MRI scans showed a 10 cm, anterior mediastinal mass with vascular encasement. The patient underwent tumor resection and radiation therapy in September 2016. Her medications at that time included lanreotide (for management of carcinoid), Lovenox (for venous thrombosis following surgery), Lexapro, ortho tri-cyclen, biotin and a daily multivitamin. She then presented to the dermatology clinic for evaluation of a lesion on the right lower chest which had been present for 10-11 months, first appearing at the time of treatment for her carcinoid tumor. She reported occasional tenderness and a slight increase in size.
Physical Examination: On exam, she was noted to have a 1.5 x 1.2 cm red, protruberant nodule on the right lateral lower chest. It was firm, slightly compressible without fluctuance or surrounding edema, induration or erythema. Radiation tattoos and other benign lesions were noted.
Histopathology: There were aggregations of epithelial cells in the dermis arranged in nests and demonstrating a trabecular pattern. Scattered mitotic figures were seen in the center of the lesion. Immunoperoxidase stains were strongly positive for both chromogranin and synaptophysin. The overall morphology and immunohistochemical staining pattern was consistent with metastatic carcinoid tumor from her thymic primary.
Clinical Course: The differential diagnosis prior to biopsy of the skin lesion included sarcoidosis, dermatofibroma, pilomatricoma or cyst. Because of its increasing size and associated symptoms, excision was planned and carried out.
Diagnosis: Atypical carcinoid tumor of thymus, metastatic to skin.
Points of Emphasis: Carcinoid tumors are derived from neuroendocrine cells and are most commonly encountered in the gastrointestinal (25%) and bronchopulmonary (65%) tracts. “Atypical carcinoid” is equivalent to a moderately differentiated neuroendocrine carcinoma which shows increased mitotic activity (2-10 mitoses per high power field), nuclear pleomorphism, or foci of necrosis. Cutaneous involvement is rare, however when present, signifies late disease and corresponds to a poorer prognosis. Cutaneous tumor deposition may be single or multiple. These nodules can appear as inflammatory, plaque-like lesions, sclerodermatous patches or as erythematous or violaceous, fixed or movable nodules. Our patient presented with a single, erythematous, well-circumscribed papule on the right lateral lower chest. The histopathology of cutaneous carcinoid tumors typically mimic those of the primary lesion. The differential diagnosis of such tumors includes primary cutaneous neuroendocrine carcinoma (such as Merkel cell carcinoma), primary cutaneous carcinoid, as well as secondary skin metastasis. Primary cutaneous carcinoid tumors are extremely rare (only 9 reported cases) and are a diagnosis of exclusion following a thorough investigation of other primary sites of neoplasia. Immunohistochemistal stains, such as CK20, may be useful in distinguishing metastatic carcinoid from Merkel cell carcinoma. In our patient, the histopathologic comparison of tumors and clinical history were consistent with a diagnosis of metastatic carcinoid from a known primary (thymus) site.
References: 1. Alwaheeb S, Ghazarian D, Boerner SL , Asa SL. Cutaneous manifestations of thyroid cancer: a report of four cases and review of the literature. J Clin Pathol 2004;57:435-8. 2. Blochin E, Stein JA , Wang NS. Atypical carcinoid metastasis to the skin. Am J Dermatopathol 2010;32:735-9. 3. Dahl PR, Brodland DG, Goellner JR , Hay ID. Thyroid carcinoma metastatic to the skin: a cutaneous manifestation of a widely disseminated malignancy. J Am Acad Dermatol 1997;36:531-7.





Case 10a: Adult-Onset Langerhans Cell Histiocytosis
Caregivers: Taylor Smith, MS-IV, Jason L. Smith, M.D., New Orleans and Rome, GA
History: An 82-year-old Caucasian male presented on Aug.15, 2017 with intense itching on the trunk and extremities and a rash that appeared approximately one year ago. The patient had seen multiple physicians in the past year and had failed courses of systemic corticosteroids, antihistamines, and topical corticosteroids. Significant past medical history included bone marrow aspiration in 2015 for work up of leukocytosis, anemia and thrombocytopenia. Bone marrow aspiration revealed a low-grade myelodysplastic syndrome.
Physical Examination: Faint, reddish-brown smooth papules (approximately 2 to 6 mm) predominantly on lower and mid back, abdomen, and flank. Few lesions noted on proximal thighs. Discrete, smooth papules coalesced and had some linearity, particularly on lower back. No mucosal lesions noted. No palpable lymphadenopathy. No hepatosplenomegaly.
Laboratory Data: Platelet count on 09/14/2015 was 35 (normal today, patient currently on Promacta 12.5 mg daily). CBC on 07/31/2017 within normal limits. Systemic work up is in progress.
Histopathology: 1) Prominent lymphohistiocytotic infiltrate with aggregates of Langerhans cells with intraepidermal abscess and interface and spongiotic changes, strongly suggestive of Langerhans cell histiocytosis; 2) Dermal plaque of epithelioid mononuclear cells/ Langerhans cells, with scattered eosinophils, and focal intraepidermal and adnexal involvement, consistent with Langerhans cell histiocytosis; 3) Aggregates of mononuclear cells admixed with lymphocytes and histiocytes, consistent with Langerhans cell histiocytosis. Intense S100 and CD1a positivity in mononuclear cells, diminished CD8 compared to CD4 & CD3. Langerin is negative. CD30 focally positive, appears reactive. GMS and AFB negative.
Clinical Course: Started on nUVB plus UVA phototherapy in office with some regression noted of lesions on trunk. Pruritus persists.
Diagnosis: Langerhans cell histiocytosis.
Points of Emphasis: Our patient had lesion morphology of monomorphic faint reddish-brown papules in clusters, few with linearity, on the trunk and proximal extremities; histopathology and immunostains were suggestive of Langerhans cell histiocytosis. Although most cases of Langerhans cell histiocytosis are seen in pediatric literature, this is a rare occurrence in adults. To date, the patient only has cutaneous involvement; work up for systemic involvement is in progress.
References: 1. Campanati A, Simonetti O, Marconi B, Giuliodori K, Ganzetti G, Brandozzi G, et al. Purely cutaneous Langerhans cell histiocytosis in an adult woman. Acta Derm Venereol. 2009;89:299–301. 2. Lian C., Lu Y., Shen S. Langerhans cell histiocytosis in adults: a case report and review of the literature. Oncotarget. 2016;7(14):18678–18683. 3. Querings K, Starz H, Balda B R. Clinical spectrum of cutaneous Langerhans cell histiocytosis mimicking various diseases. Acta Derm Venereol. 2006;86:39–43.







Case 10b: Langerhans Cell Sarcoma
Caregivers: Jacqueline Bucher, M.D., Holly H. Volz, M.D., Thomas Davis, M.D., San Antonio, TX
History: A 66-year-old female presented to clinic for evaluation of a tender, enlarging growth on the left calf, present for 2 months. Her past medical history was significant for a recent diagnosis of B-cell chronic lymphocytic leukemia (CLL) with a 17p chromosomal deletion and a remote history of breast cancer.
Physical Examination: Physical examination revealed a 1-cm, tender, violaceous papule on the left posterior calf. The remainder of the physical exam was unremarkable.
Histopathology: A shave biopsy from the left calf demonstrated a dense nodular infiltrate of atypical mononuclear cells with moderate pleomorphism in the dermis, many with an epithelioid appearance. Numerous mitotic figures were present within the infiltrate, and there were also many extravasated erythrocytes within the surrounding dermis. A panel of IHC stains revealed positive staining of neoplastic cells with S100, CD1a, and CD31. Ki-67 revealed a proliferation rate of 30-40%. The neoplastic cells were negative for CD30, CD34, SOX10, Melan-A, and p63.
Clinical Course: PET/CT scan showed diffuse lymphadenopathy; however, there was no evidence of local regional lymphadenopathy on the left leg, near the site of the lesion. Imaging guided biopsy from the retroperitoneal lymph nodes was consistent with her known history of leukemia. Soon after initial skin biopsy, the patient developed several new, tender lesions on her left postauricular scalp, frontal scalp, and right thigh. Shave biopsies of these new lesions revealed similar findings to the initial biopsy. The patient was subsequently started on chemotherapy with R-EPOCH.
Diagnosis: Langerhans cell sarcoma
Points of Emphasis: Langerhans Cell Sarcoma (LCS) is an extremely rare proliferative disorder of Langerhans cells with an aggressive clinical course, often involving multiple sites, including the skin, lymph nodes, liver, spleen, lung and bone. LCS may arise de novo, progress from Langerhans Cell Histiocytosis (LCH), or arise in a setting of a myeloproliferative or lymphoproliferative disease. LCS shares many features with its counterpart, Langerhans Cell Histiocytosis (LCH), including positivity for CD4, CD1a, S100, and Birbeck granules stained with langerin. It may be differentiated from LCH by the degree of cytologic atypia, elevated mitotic rate, and clinical aggressiveness. As in our case, CD31 positive staining has been reported in LCS and LCH, but is not present on normal epidermal Langerhans cells. CD31 is commonly used as an endothelial marker; however, it may also be seen on monocytes, platelets, neutrophils, and subsets of T-cells. It is hypothesized that the upregulation of CD31 on neoplastic Langerhans cells contributes to their migratory capacity. Our patient is the third reported case in the literature with concurrent LCS and CLL/SLL (small lymphocytic lymphoma). Recent studies have demonstrated the common clonal origin of both histiocytic/dendritic sarcomas and lymphoproliferative diseases arising in the same individual. Two hypotheses exist as to why this newly recognized phenomenon, termed “transdifferentiation”, occurs. The first involves the direct transdifferentiation of neoplastic B cells to malignant dendritic cells. The second implies multiple steps involving de-differentiation of neoplastic lymphocytes to early progenitor cells followed by re-differentiation to dendritic cells. The 17p deletion in our patient contains the TP53 gene. Patients with this mutation show marked resistance to chemotherapies and generally have a poorer prognosis. This mutation is significant because it may be a potential risk factor for the transformation of CLL/SLL. One publication to date investigates the cytogenetics of dendritic/histiocytic neoplasms arising from SLL/CLL. The 17p mutation was detected in all six dendritic/histiocytic neoplasms and two cases of the corresponding CLL/SLL. In summary, this case demonstrates the rare phenomenon of transdifferentiation from CLL to LCS and provides further evidence for the association of this process with the 17p deletion.
References: 1. Nakamine, H. Langerhans Cell Histiocytosis and Langerhans Cell Sarcoma: Current Understanding and Differential Diagnosis. J Clin Exp Hematop. 2016 Dec; 56(2): 109-118. 2. Weiwei Chen, Ronald Jaffe. Langerhans Cell Sarcoma Arising from Chronic Lymphocytic Lymphoma/Small Lymphocytic Leukemia: Lineage Analysis and BRAF V600E Mutation Study. N Am J Med Sci. 2013 Jun; 5(6): 386-391. 3. Shao H. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol. 2011 Nov;24(11):1421-32.






Case 11: Clear Cell Sarcoma of the Large Toe
Caregivers: John Millns, M.D., Terrance Hopkins, M.D., Michael Heaphy, M.D., Tampa & Brandenton, FL
History: An 80-year-old female presented with a 50 year history of a 1-cm nodule on the left large toe near the MTP joint which had recently began changing over the past nine months (increasing in size, nodularity, and erythema). The patient was in otherwise good health with no other major complaints or underlying illnesses.
Physical Examination: The patient was found to have a 1-cm, pink, slightly mobile nodule on the dorsal MTP joint of the left large toe. No evidence of lymphadenopathy was identified in the popliteal fossa or groin region.
Laboratory Data: CBC with differential - normal; metabolic panel - normal; chest x-ray- normal; MRI of the head- negative. Molecular analysis was completed at Texas Children’s Hospital. In this analysis, a EWSR1/CREB1 fusion transcript was detected. This molecular testing result provides confirmation of the diagnosis of clear cell carcinoma (melanoma of soft parts).
Histopathology: Skin and subcutis infiltrated by an atypical spindle cell neoplasm composed of fusiform cells possessing ovoid nuclei with prominent nuclei and a modest amount of amphophilic cytoplasm. Lesional cells are arranged in compact nests and short fascicles separated by a hyalinized stroma. Scattered mitotic figures are identified. HMB-45 immunoperoxidase stain was diffusely positive.
Clinical Course: The patient underwent wide excision with 1-cm margins of the involved lesion with excellent healing response. Repeat ultrasound of the popliteal fossa and inguinal region has been negative.
Diagnosis: Clear cell sarcoma
Points of Emphasis: Clear cell sarcoma (melanoma of the soft parts) is a rare tumor that usually arises in young adults and shows a predilection for females and occurs most often in the distal extremities, particularly the hands and feet, as noted in this case. This tumor is prone to local recurrence and widespread metastases, often many years after the primary diagnosis. In spite of the somewhat broad differential diagnosis, melanoma remains the most difficult simulator of clear cell sarcoma. About 60% of the cases contain variable amounts of melanin pigment which can be highlighted by special stains. Tumor cells are positive with S-100 protein, HMB-45, Melan-A, and NSE. In this case, an EWSR1/CREB1 fusion transcript associated with t(2;22) and characteristic of a subset of clear cell sarcomas was detected in the tissue sections differentiating clear cell sarcoma from melanoma.
References: 1. Mavrogenis A, Bianchi G, Stavropoulos N, Papagelopoulos P, Ruggieri P. Clinicopathologic features, diagnosis and treatment of clear cell carcinoma/melanoma of soft parts. Hippokratia. 2013 Oct;17(4):298-302. 2. Fusumae T, Kamiva K, Maekawa T, Komine M, Murata S, Ohtsuki M. Clear cell sarcoma with intraepidermal nests requiring the differential diagnosis of malignant melanoma. J Dermatol. 2017 Mar 33. Bombonato C, Moscarella E, Longo C, Castagnetti F, Nicoli D, Piana S. Subcutaneous pigmented clear cell sarcoma as a challenging simulator of melanoma. Australas J Dermatol. 2017 Aug 17.












Case 12: Angiosarcoma at the site of a previous dialysis Graft
Caregivers: Michele Zerah, M.D., Elizabeth Dugan, M.D., Washington D.C.
History: A 65-year-old male presented for evaluation of severe pain in his left upper extremity at the site of a previous dialysis graft. He had a 7-year history of localized pain at the graft site; the graft had been previously removed at an outside hospital. He developed ulceration at the site with persistent bleeding after multiple debridements. Patient denied loss of sensation or motor strength but reported that he was not able to use his extremity due to intractable pain. He had no known history of immunosuppression.
Physical Examination: On initial examination, the patient had an 8 x 5 “lazy-S” shaped ulcer just superior to the left antecubital fossa with granulation tissue and foci of punctate hemorrhages. The ulcer had rolled, gray borders. No cervical lymphadenopathy or peripheral edema was noted. At the time of biopsy three weeks later, the size of the ulcer had increased to 11.1 x 7.5 cm.
Laboratory Data: WBC- 5.2; Hgb- 6.9; Hct- 22.3; Plt- 248; MCV- 99.6; Diff- WNL; INR- 1.2; HIV- negative (4/2014); HCV, HBV, and HAV- negative (7/2017); superficial bacterial culture: Enterobacter cloacae.
Histopathology: Sheets of large, plump atypical epithelioid cells associated with extravasated RBCs and hemosiderin. Tumor extended from the deep dermis into the deep soft tissue. Individual cells demonstrated enlarged nuclei with multiple prominent nucleoli and scattered mitoses. Poorly formed vascular spaces demonstrated a dissecting growth pattern and intracytoplasmic microluminae. Some larger vascular spaces contained frond-like infoldings lined by plump, atypical cells. Antibodies to CD31, CD34 and AE1/AE3 showed positive staining. Antibodies to S-100, Melan-A, CK-7, CK20, HHV-8 and CEA were negative.
Clinical Course: After 2 weeks of treatment with levofloxacin for superinfection of the wound, the patient underwent left upper extremity amputation. A CT scan of the chest, abdomen and pelvis, a PET scan, and an MRI of the brain were negative for metastases. Sentinel lymph node biopsy was also negative.
Diagnosis: Epithelioid angiosarcoma at the site of a former arteriovenous graft site in an immunocompetent host
Points of Emphasis: This is a rare diagnosis of epithelioid angioarcoma at a former hemodialysis site. This is regarded as an aggressive tumor and average survival is approximately 9 months. Few cases have been reported without immosuppression and most at fistula sites.
References: 1. Osrochi Y, Razi K, Stebbing J, Crane J. Angiosarcoma and dialysis-related arteriovenous fistulae: a comprehensive review. Eur J Vasc Endovasc Surg 2016; 51:127-133. 2. Hart J, Mandavilli S. Epithelioid Angiosarcoma. Arch Pathol Lab Med 2011;Vol 135: 268-272.










Case 13: Pleomorphic Fibroma
Caregivers: Taylor Dickerson, B.S., Aimee Coscarart, M.D., Cole Claiborne, M.D., Pamela Martin, M.D., New Orleans, LA
History: A 32-year-old Caucasian male with history of embryonal testicular cancer and seminoma, status post orchiectomy and chemotherapy, presented with a six-month history of an asymptomatic “bump” on his left arm.
Physical Examination: 1.1 cm faintly pink, soft tissue tumor in the left lateral antecubital fossa
Laboratory Data: None
Histopathology: Skin with an ulcerative defect which was lined on the dermal side by organizing fibroplasia and a pigmented histiocytic reaction. Subjacent dermis showed a proliferation of atypical spindled cells, some multinucleated, and some isolated between fibrocytes. No expansile or tumoral pattern was seen but the lesional cells were present interstitially. No significant mitotic activity seen. The lesional cells showed slight CD34 positivity. CD163 was also weakly positive. SOX10 and S100 immunostains were negative.
Clinical Course: The lesion was subsequently excised. The patient will be followed for recurrence.
Diagnosis: Pleomorphic fibroma
Points of Emphasis: Pleomorphic fibroma (PF) is a benign, polypoid, or dome-shaped cutaneous neoplasm with cytologically atypical fibrohistiocytic cells. Clinically it may resemble a nevus, neurofibroma, hemangioma, fibrokeratoma, or fibroepithelial polyp. Histological differential includes: atypical fibroxanthoma, dermatofibroma with monster giant cells, giant cell fibroblastoma, desmoplastic Spitz nevus, and desmoplastic melanoma. It does not appear that this is a sequela of chemotherapy or metastasis from embryonal carcinoma or seminoma.
References: 1. Nakamura, Y., Nakamura A., Muto, M. A case of pleomorphic fibroma of the skin presenting as intradermal nodule. Am J Dermatopathol. 2015 Feb; 37(2): 175. 2. Yadave, Y. et al. Cytomorphology of pleomorphic fibroma of skin: A diagnostic enigma. J Cytol. 2013 Jan;30(1):71.







Case 14: Plexiform Neurilemmoma Arising Near a Surgical Scar
Caregivers: Taylor Smith, MS IV, Jordan Brooks, M.D., W.C. Cole Claiborne, M.D., New Orleans, LA
History: 73-year-old female presents with a 2-year history of a slow-growing asymptomatic lesion on her left lower extremity. She denies any similar lesions elsewhere. Other medical history: leg cramping, neuropathy, morbid obesity, pain in joint involving lower leg, and hypothyroidism. Surgical history includes total knee replacement. The patient denied any personal or family history of brain tumors or hearing loss.
Physical Examination: 2.1 cm firm, exophytic white/yellow nodule on left medial knee, medial to surgical scar overlying joint.
Histopathology: Skin biopsy of the left medial knee showed a tumor composed of multiple, well-circumscribed nodules of spindle cells arranged in short, intersecting fascicles. The elongated nuclei, some which were curved, showed fine chromatin. The surrounding stroma was myxoid. There was no significant cytologic atypia, mitotic activity, or necrosis. Immunoperoxidase stains were positive for S100 protein and negative for actin and pan melanocytic cocktail (HMB-45, MART-1, and tyrosinase). A small percentage of cells were positive for desmin. The proliferation rate estimated from the nuclear labeling index of Ki-67 was low and approximately 2%. This lesion should be completely removed. The lesion extends to peripheral and deep histologic margins.
Clinical Course: Monitoring clinically. The patient declined surgical excision.
Diagnosis: Plexiform neurilemmoma
Points of Emphasis: This patient, with no personal or family history of neurofibromatosis or schwannomatosis, presented with a solitary plexiform neurilemmoma overlying a surgical scar. Schwannomas are known to be focally desmin positive in a minority of cases. I feel the constellation of morphological and immunohistochemical findings are characteristic of plexiform schwannoma. This type of schwannoma is known to be associated with NF2 and schwannomatosis in a very small percentage of cases. To our knowledge there are no known cases of schwannomas arising within surgical sites or within surgical scars. Close proximity to total knee replacement surgical scar is likely coincidental.
References: 1. Berg J C, Scheithauer B W, Spinner R J, Allen C M, Koutlas I G. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39(5):633–640. 2. Hébert-Blouin M N, Amrami K K, Scheithauer B W, Spinner R J. Multinodular/plexiform (multifascicular) schwannomas of major peripheral nerves: an underrecognized part of the spectrum of schwannomas. J Neurosurg. 2010;112(2):372–382.







Case 15: Atypical (Symplastic) Piloleiomyoma with Follicular Induction
Caregivers: Caroline Hagan, MS-III, Shreveport, LA
History: A 58-year-old white male presented to clinic with a firm, 0.8 x 0.6 cm pink-brown, dome-shaped papule on the left shoulder. The patient had past medical history of diabetes mellitus, hypertension, and hyperlipidemia and was being treated with atorvastatin, fenofibrate, lisinopril, metoprolol, Xigud, and tanzeum. The patient also had vitiligo on the fingertips and volar wrist but denied any personal or family history of other skin conditions. The patient had no known drug or environmental allergies, denied tobacco use and street drugs, and endorsed occasional alcohol use.
Physical Examination: Initial presentation showed a single, symmetrical 0.8 x 0.6 cm well-circumscribed, round, pink-brown, dome-shaped papule on the left shoulder with no surrounding erythema, swelling, or tenderness to palpation. The lesion appeared mildly hyperpigmented circumferentially but was otherwise uniformly pigmented and not ulcerated. A 0.9 x 0.8 x 0.2 cm shave biopsy was obtained.
Laboratory Data: No significant lab values were obtained.
Histopathology: H&E stained histologic sections demonstrated a raised lesion with irregular, verruciform epidermal acanthosis and hyperkeratosis. Evidence of hair follicle differentiation in the form of sebaceous glands and papillary mesenchymal bodies was noted in the epidermis. The underlying papillary dermis was largely replaced by cellular, interlacing fascicles of eosinophilic spindle cells with ovoid to cigar-shaped, variably hyperchromatic nuclei and perinuclear vacuoles. The proliferation extensively involved the reticular dermis and extended to involve the superficial subcutis. The mitotic rate was noted as less than 1 per 10 HPF. No atypical mitotic figures or necrosis were identified. Special stains included: desmin – positive (4+ cytoplasmic), SMA- positive (4+ cytoplasmic), and factor 13a - negative.
Clinical Course: Histopathologic examination revealed an atypical piloleiomyoma with unusually increased cellularity; a low grade or partially sampled leiomyosarcoma could not be excluded. The pathologist suggested a complete excision for full histologic study. The excision specimen showed residual piloleiomyoma with clear margins. Follow-up visit one month later showed no signs of recurrence. No additional lesions were noted on the trunk or extremities. Close follow-up was recommended.
Diagnosis: Atypical (symplastic) piloleiomyoma with follicular induction
Points of Emphasis: To our knowledge, follicular or sebaceous induction has not been previously reported in association with a smooth muscle neoplasm. Recent studies suggest this phenomenon may be mediated by complex interactions between the Wnt family of signaling molecules and beta-catenin. Atypical (symplastic) piloleiomyoma may in some cases represent a low grade form of leiomyosarcoma. Given the uncertain biologic potential, complete excision of atypical leiomyomas is recommended to prevent possible recurrence.
References: 1. Yung A, et al. J Cutan Pathol 2008; 35(3): 329-31. 2. Fu J, Hsu W. J Invest Dermatol 2013; 133(4): 890-898. 3. Miettinen M. Modern Pathology 2014; 27: S17-S29.




Case 16: Dystrophic Calcinosis Cutis
Caregivers: Zachary Gillooly, M.D., Jacqueline Bucher, M.D., Robert Gilson, M.D., San Antonio, TX
History: A 59-year-old Hispanic female on oral tacrolimus immunosuppression was admitted to a tertiary care hospital from an outside hospital with community-acquired pneumonia and suspected cellulitis of the left posterior neck and shoulder. Past medical history was significant for adenocystic carcinoma of the left submandibular gland (1983; status post excision and radiation therapy) as well as non-alcoholic steatohepatitis complicated by hepatocellular carcinoma (orthotopic liver transplant performed in 2015). On admission, the lesions on the patient’s left posterior neck and shoulder were very firm, erythematous, and tender and had been present for at least three months; some were chalky white. The patient had a prior biopsy performed at an outside otolaryngology clinic, in addition to oral corticosteroid therapy and intralesional corticosteroid injections, with no relief from the pain. Prior CT imaging from the outside hospital showed “tumoral calcinosis as well as skin thickening and subcutaneous inflammation concerning for cellulitis without evidence of abscess.” MRI showed cervical stenosis and intraspinal calcification at the level of C4-C5 with paraspinal muscle calcification.
Physical Examination: Cutaneous lesions were localized to the left upper posterior neck and shoulder. A single, 8 cm, discrete, indurated, hyperpigmented, erythematous, and sclerotic plaque was present, with smaller 0.5- 1.0 cm central, chalky white, firm deposits extruding from the skin. No associated warmth or purulent drainage was appreciated. The oral mucosa was clear, and there was no lymphadenopathy present in the neck or axillae. Focal, left hand neurologic deficits were noted. A posterior neck core biopsy was performed under ultrasound guidance.
Laboratory Data: The patient’s serum calcium, phosphorus, and parathyroid hormone levels were within normal limits. Her renal function was also within normal limits. The patient had a mild leukocytosis at the time of admission.
Histopathology: H&E stained sections showed fibroadipose tissue with fibrosed and atrophic underlying skeletal muscle with abundant calcified and amorphous material. Focal chronic inflammation was identified. There was no acute inflammation or evidence of malignancy.
Clinical Course: At the outside hospital, the patient was treated with cefepime and azithromycin for her community-acquired pneumonia and concern for a skin/soft tissue infection. The patient was transferred to a tertiary care hospital for suspected spinal cord impingement and transitioned to levofloxacin. The erythema overlying her left posterior neck and shoulder appeared to improve on antibiotics, but there was no change in the indurated and sclerotic plaque or the chalky white lesions. Importantly, there was no change in the patient’s level of pain. Given the CT and MRI findings on admission with intraspinal calcification, cervical spine stenosis, and paraspinal muscle calcification, otolaryngology and neurosurgery were both consulted. No urgent surgical intervention was planned. From a dermatologic standpoint, the patient was started on topical triamcinolone 0.1% ointment, twice daily, in addition to minocycline 50mg once daily as an anti-inflammatory agent. Additionally, the patient had no systemic or cutaneous findings consistent with CREST syndrome or other connective tissue diseases, including dermatomyositis.
Diagnosis: Dystrophic calcinosis cutis
Points of Emphasis: Dystrophic calcinosis cutis is the most common subtype of calcinosis cutis and results from the abnormal deposition of insoluble calcium salts at sites of previously damaged soft tissue. The condition is most commonly associated with an underlying connective tissue disorder but several case reports have been published of dystrophic calcinosis cutis arising at sites of previous radiation therapy. Calcinosis cutis tends to occur as a late complication of radiation therapy in these patients. Management of calcinosis cutis is often challenging and suboptimal. The evidence behind these therapeutic options is generally only from case reports or case series at best. Calcium channel blockers, namely diltiazem, are a common first-line therapy for most patients with calcinosis cutis. However, given the drug-drug interactions between immunosuppressants such as tacrolimus and calcium channel blockers, diltiazem was not an option in our patient. Anti-inflammatory agents, such as colchicine and minocycline, have also been used for calcinosis cutis, especially in cases presenting with ulceration. In a case series of nine patients treated with 50-100mg of minocycline once daily, eight showed definite improvement. The earliest sign of improvement occurred at one month of therapy, but the mean length of therapy was 3.5 years. Additional therapies include bisphosphonates and sodium thiosulfate (topical, intralesional, and intravenous), as well as intravenous immunoglobulin and infliximab. Lastly, surgical excision is sometimes an option in selected patients.
References: 1. Lee HW, Oh SH, Chang SE, Lee MW, Choi JH, Moon KC, Koh JK. Dystrophic calcinosis cutis following chest wall and breast irradiation. The Journal of Dermatology. 2005;32:1055-1057. 2. Robertson LP, Marshall RW, Hickling P. Treatment of cutaneous calcinosis in limited systemic sclerosis with minocycline. Annals of Rheumatic Diseases. 2003;62(3):267.





Case 17: Osteoma Cutis with Gelatinous Transformation in a patient with rapid weight loss
Caregivers: Cynthia Griffith, PA-C, Melissa Dillard, M.D., Danielle de Stefano, M.D., Elena Maxim, MS-IV, Travis Vandergriff, M.D., Dallas, TX
History: A 45-year-old woman, with history of a 200-lb weight loss in two years, presented to dermatology with “small bumps under the skin” of several years duration. Her weight loss was intentional and attributed to a gastric sleeve procedure performed in 2015. The “bumps” were not associated with any itching or pain. Additional surgical history included a cholecystectomy, a hysterectomy, and a nephrectomy for unknown reason. Her only medication at the time of presentation was conjugated estrogen cream.
Physical Examination: The patient was a well-developed, fully alert female. A full physical examination, including buttocks and genitalia was performed. Multiple, subcutaneous nodules, each measuring < 5 mm2 were observed on the body: right upper arm x 2, left upper arm x 1, and left inguinal fold x 1. The majority of the lesions were soft and mobile with no obvious overlying skin changes, clinically most consistent with a lipoma or cyst. The lesion on the posterior right upper arm was similarly small but firm to palpation. Two punch biopsies of the right upper arm were performed: Site A (right upper arm anterior) and site B (right upper arm posterior).
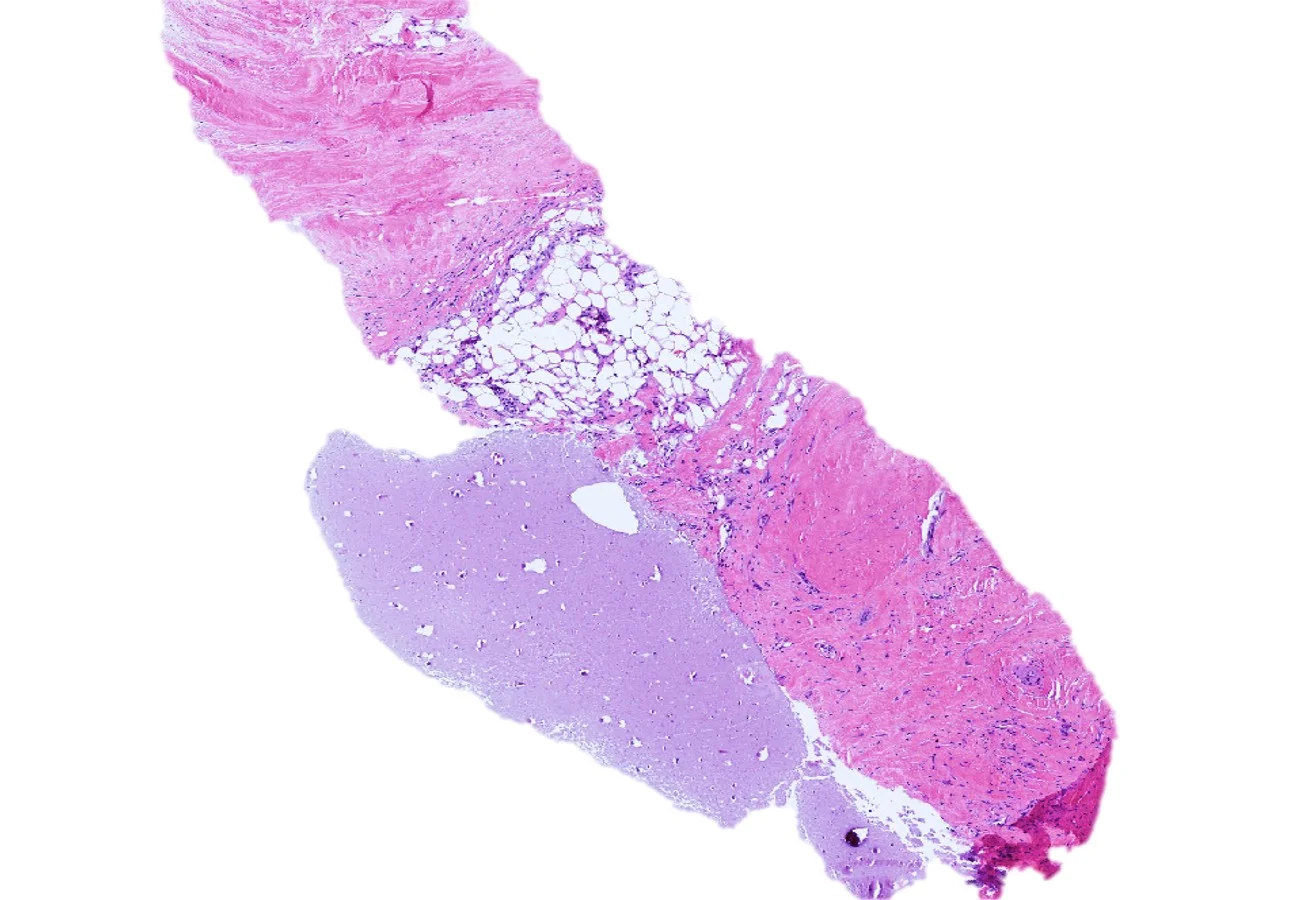
Histopathology: Sections of site A showed circumscribed lobules of adipose tissue containing a few blood vessels and fibrous strands, consistent with a lipoma (not pictured). Site B showed a nodular lesion in the dermis composed of a peripheral rim of bony spicules with lacunae and calcification in a pattern typical of osteoma cutis. Centrally, there was atrophic fat and scattered lymphocytes contained within a diffuse, gelatinous matrix.
Clinical Course: The patient reported some mild bleeding following biopsy and has since been lost to follow-up.
Diagnosis: Osteoma cutis with gelationous transformation
Points of Emphasis: Gelatinous transformation of bone marrow is a condition characterized histologically by fat cell atrophy, hematopoietic cell hypoplasia, and deposition of extracellular, gelatinous material. The gelatinous material, usually amorphous and slightly granular, is comprised of acid mucopolysaccharides (hyaluronic acid) which show positive staining with Alcian blue. Older but synonymous terms for gelatinous transformation of marrow include “serous atrophy of fat” and “starvation marrow.” A rare entity, gelatinous transformation of bone marrow has been observed in both animals and humans in extreme starvation states, such as those seen with severe malnutrition, cachexia and anorexia nervosa. This finding has also been observed in association with acute febrile illness, in viral illness, such as AIDS, and in alcoholism. Although the pathogenesis is unclear, it is thought to be the result of basic bioregulatory processes where ground substance is a replacement for adipocytes used in catabolism and hematopoietic cell loss is secondary to an unfavorable marrow microenvironment. Here we report an unusual case of gelatinous transformation, not within bone marrow but rather within a cutaneous lesion, an osteoma cutis. An initial review of the medical literature has revealed no such similar reported cases. Although a rare pathologic finding, we want to emphasize the importance of this entity in practice as it implies severe underlying illness. Particularly in our case, the finding is likely related to severe malnutrition in the setting of extreme weight loss. Additional laboratory workup and testing may be warranted for these patients.
References: 1. Bain BJ, Clark DM, Lampert IA, Wilkins BS. 2001. Bone Marrow Pathology, 3rd ed. Blackwell, Ames, IA, 90-140. 2. Böhm J. 2000. Gelatinous transformation of the bone marrow - the spectrum of underlying diseases. Am J Surg Pathol 24:56-65. 3. Travlos GS. 2006. Histopathology of the bone marrow. Toxicol Pathol 34:566-598.





Case 18: Pigmented Fungiform Papillae of the Tongue: An Incidental Finding in a Patient with Mycosis Fungoides
Caregivers: Heather Wickless, M.D., Melissa Dillard, M.D., Danielle de Stefano, M.D., Elena Maxim, MS-IV, Travis Vandergriff, M.D., Dallas, TX
History: A 28-year-old Hispanic male with history of Stage IA mycosis fungoides presented with “new dark spots on the tongue.” He denied pain or pruritus. He also denied ever having taken minocycline or any prescribed medications. He did endorse daily intake of omega 3 and a multivitamin. Past dermatologic diagnoses included a halo nevus, generalized xerosis treated with Cetaphil, and idiopathic guttate hypomelanosis on the back. At the current visit, he denied systemic symptoms. His mycosis fungoides had been clinically stable for the past five years with home, narrowband UVB light phototherapy twice a month.
Physical Examination: Physical examination of the oral cavity was significant for multiple dark-brown structures on the distal tongue. Dermoscopic evaluation showed several fungiform papillae with pigmented borders in a rose petal pattern. No other unusual hyperpigmented lesions were seen on the body.
Histopathology: A shave biopsy of tongue showed papillae with melanophages in the lamina propria.
Clinical Course: The patient was reassured of benign etiology of his tongue lesion.
Diagnosis: Pigmented fungiform papillae of the tongue
Points of Emphasis: Normal fungiform papillae of the tongue are pink “mushroom-shaped” projections predominantly located on the lingual margin but also irregularly distributed on the dorsal surface of the tongue. Pigmented fungiform papillae (PFP) are considered a normal variant with increased pigmentation. The finding is more prevalent in individuals with darker skin but the pathogenesis remains unclear. Other pigmented lesions of the oral mucosa include Peutz-Jeghers syndrome, hemochromatosis, Addison’s disease, black hairy tongue, melanocytic nevi and melanoma. Multiple medications and exogenous substances such as amalgam tattoo, anti-malarials and antibiotics can also cause mucosal pigmentation. While some of these entities can be differentiated from PFP on clinical grounds alone, dermoscopic patterns can be extremely helpful in distinguishing this entity from other melanocytic conditions. Benign lesions can show one or several patterns with a regular distribution, while melanomas tend to have several patterns which are irregularly distributed. A rose petal pattern is more consistent with PFP. In our case, dermoscopy was a helpful tool in distinguishing possible melanocytic oral processes from this uncommon normal variant of lingual papillae. Its use should be encouraged as part of a detailed physical examination of the oral mucosa in similar situations, in order to avoid incorrect diagnoses and unnecessary testing. Microscopic studies could be performed in cases of remaining diagnostic uncertainty.
References: 1. Holzwanger J.M., Rudolph R.I., an Heaton C.L.: Pigmented fungiform papillae of the tongue: A common variant of oral pigmentation. Int J Dermatol 1974; 13: pp.403-408. 2. Pinos-Leon, V., Dermoscopic Features of Pigmented Fungiform Papillae of the Tongue. Dermatology. 2015. Volume 106, Issue 7, pp 593-594. 3. Marcoval, J, et al. Pigmentacion de las papilas fungiformes linguales. A proposito de dos casos. Actas Dermosifiliogr. 2011; 102: pp 739-740.






Case 19: Massive Localized Edema of the Vulva Mimicking a Neoplasm
Caregivers: Melissa Dillard, M.D., Danielle de Stefano, M.D., Travis Vandergriff, M.D., Elena Maxim, MS-IV, Dallas, TX
History: A 38-year-old, morbidly obese female was referred for an oncology consultation due to massive enlargement of the vulvar region. The enlargement was first noticed by the patient 6 years ago, and since onset, had doubled in size. The lesion was occasionally painful and limited sexual activity. Her past medical history included hypertension, endometriosis, and a cervical excision procedure at age 18 for an abnormal pap smear. Familial cancer history included lung cancer in both an uncle and a grandfather and pancreatic cancer in her grandmother.
Physical Examination: A massively enlarged, verrucous/plaque-like lesion with multiple, small coalescing polypoid masses were seen encompassing the vulvar area. The lesions extended down to the anus. Similar but smaller plaques were also noted on the labia majora. The mons pubis contained a separate, 8 x 12 cm, firm but mobile, pedunculated mass.
Laboratory data: A pelvic MRI was requested but not performed due to financial reasons.
Histopathology: Papillomatosis, hyperkeratosis, and dermal fibroplasia with dilated lymphatics in a circumscriptive pattern was observed. There was no evidence of malignancy.
Clinical Course: The patient underwent a partial vulvectomy with excision of the mons pubis mass. After 5 weeks, the patient reported significant improvement in her quality of life with reduction in pain and a new ability to exercise.
Diagnosis: Massive localized edema of the vulva
Points of Emphasis: Massive localized lymphedema of the vulva is a rare, “pseudosarcoma” associated with obesity, immobilization, prior surgery, and blunt trauma. It is characterized by prominent expansion of the vulvar connective tissue leading to massive, pedunculated masses and verrucous, plaque-like lesions of the skin. These lesions can mimic cancers such as lymphangiosarcoma. Although rare, this is an important entity to recognize due to its gross similarities to a neoplasm. In addition, because a significant proportion of angiosarcomas can arise in the setting of chronic lymphedema, close follow-up is recommended in these cases.
References: 1. Heller, Debra et al. Massive Localized Lymphedema of the Vulva: Report of a Case and Review of the Literature. Journal of Lower Genital Tract Disease. April 2014. Vol 8, Issue 2, pp E46-E49. 2. Habibe, Kurt, et al. Massive Localized lymphedema: a clinicopathologic study of 46 patients with an enrichment for multiplicity. Modern Pathology (2016) 29, pp 75-82.





Case 20: Anaplastic Plasmacytoma
Caregivers: Marisa Belaidi, M.D., Kathryn Olivier, MS-IV, Pamela Martin, M.D., Elizabeth Grieshaber, M.D., New Orleans, LA
History: A 93-year-old Caucasian woman presented for evaluation of a “spot” on her left upper arm which had been persistent for at least several weeks. The lesion was asymptomatic, did not bleed, and had not been treated in the past. No history of trauma to the area was elucidated. The patient denied history of any similar lesions elsewhere on her body. A review of systems was negative for systemic complaints.
Physical Examination: Left lateral upper arm: solitary, 1-cm, well-circumscribed, smooth red plaque with a surrounding patch of erythema and scaling; no other similar lesions were found.
Lymph Nodes: within normal limits
Histopathology: The shave biopsy showed complete loss of epithelium. A proliferation of vascular structures was present in an inflamed stroma. Most notably, immature plasma cells, with enlarged nuclei, prominent nucleoli and a blastic appearance, were identified. These cells were CD138, MUM1, and CD79a positive and stained focally with CD20. EBV and HHV-8 were negative. Kappa/lambda stains showed the presence of both light chains with an increased number of lambda-positive cells, but the blastic cells showed kappa predominance. Ki-67 showed a markedly increased percentage of staining cells. Background cells stained with CD4 and CD5. FISH studies showed an RB1 (13q14) deletion. The lesion extended to the margins.
Clinical Course: The patient was hospitalized for a hip fracture shortly after biopsy. Workup for underlying systemic myeloproliferative disorder was negative and the hip fracture was determined to be an isolated, unrelated condition following a fall. She presented to clinic two months following her initial biopsy at which time the lesion was no longer present.
Diagnosis: Anaplastic plasmacytoma
Points of Emphasis: The term extramedullary plasmacytoma (EMP) refers to a proliferation of clonally expanded plasma cells present outside of the bone marrow. Though the upper aerodigestive tract is most commonly affected, EMP has been reported at multiple other sites including the skin, in which case it is called “cutaneous plasmacytoma.” EMP can be divided into two pathophysiologic subsets that are clinically and histologically indistinguishable: primary EMP, which occurs independently of underlying plasma cell myeloma (PCM), and secondary EMP, which presents as either a metastasis or direct extension of an underlying PCM. Clinically, both primary and secondary cutaneous plasmacytomas present as violaceous, smooth, dome-shaped nodules most commonly seen on the face, trunk, and extremities. Histology demonstrates an infiltration of atypical plasma cells (CD138+) with increased mitotic activity and extension into the reticular dermis and/or subcutis. In situ hybridization reveals clonal kappa or lambda light chains. While no correlation between specific chromosomal abnormalities and clinical features has been identified, genetic analysis has suggested that primary EMP and PCM represent distinct entities. It is important to differentiate the two variants, as they portend different prognoses and treatment courses. Primary cutaneous plasmacytoma (PCP) has been reported to indicate a more favorable prognosis, especially in the setting of solitary lesions which can be treated with either simple excision or local radiation. The differential diagnosis of PCP includes secondary cutaneous plasmacytoma, other primary B-cell lymphomas such as plasmablastic lymphoma (PBL), and several infectious processes such as syphilis or Borrelia burgdorferi. Our case expressed overlapping features of both PCP and PBL. Differentiating the two was of key importance, as PBL tends to follow an aggressive clinical course with most experiencing poor outcomes despite early intervention. PBL is commonly associated with HIV infection, however it has been seen in other immunodeficient settings including iatrogenic immunosuppression and in elderly patients. It may be associated with EBV and HHV-8, for which our specimen tested negative. The final diagnosis of primary cutaneous anaplastic plasmacytoma was predicated upon the overall clinical and histopathologic picture combined with extensive literature review.
References: 1. Comfere NI et al. Cutaneous extramedullary plasmacytoma: clinical, prognostic, and interphase cytogenetic analysis. Am J Dermatopathol. 2013;35(3):357-63. 2. Muscardin LM et al. Primary cutaneous plasmacytoma: report of a case with review of the literature. J Am Acad Dermatol. 2000;43(5 Pt 2):962-5. 3. Kazakov DV et al. Primary cutaneous plasmacytoma: a clinicopathological study of two cases with a long-term follow-up and review of the literature. J Cutan Pathol 2002:29:244–248.





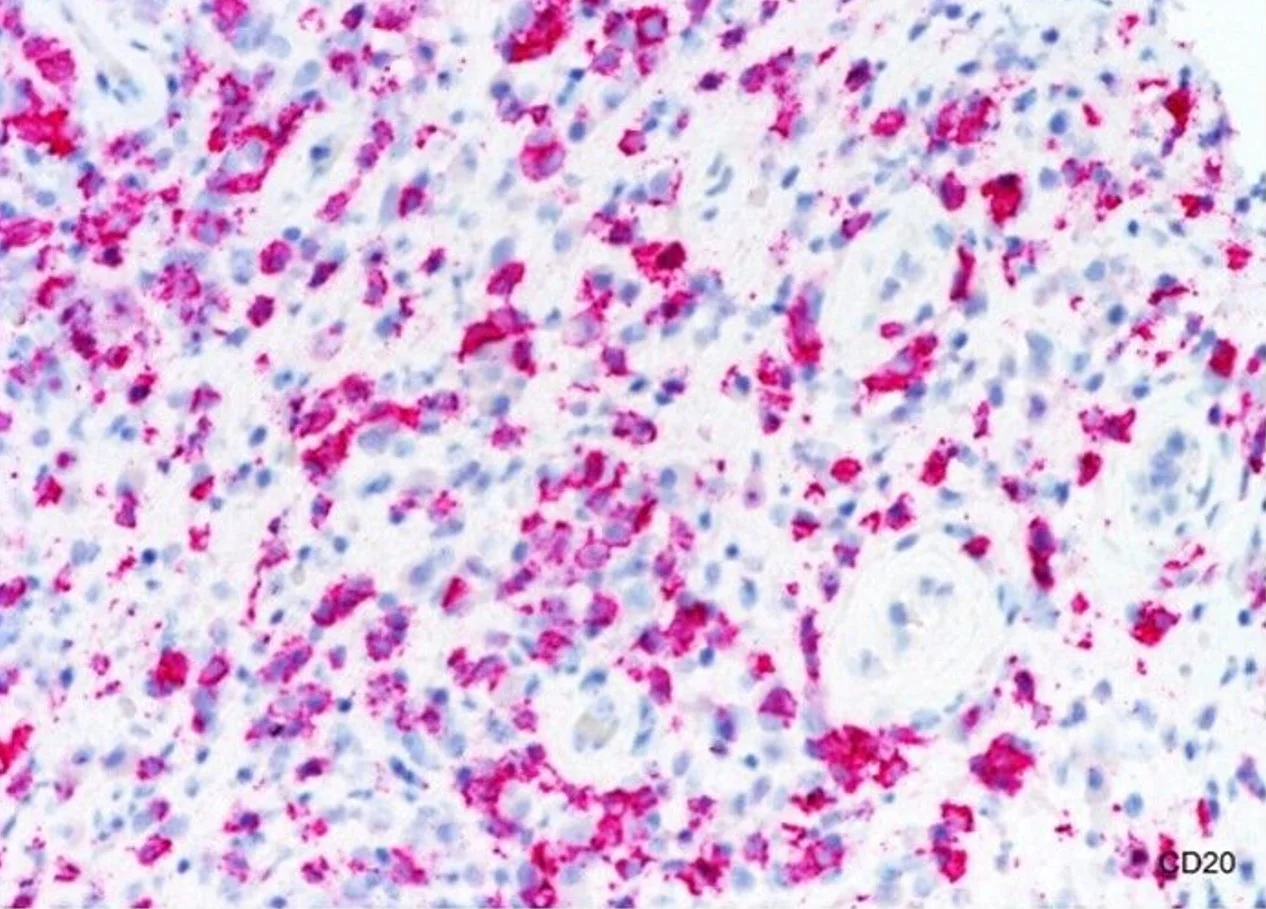





Case 21: Angioinvasive Lymphomatoid papulosis
Caregivers: Taylor Smith, MS-III, Jason L. Smith, M.D., New Orleans, LA and Rome, GA
History: A 57-year-old white female presented with several, oval-shaped, pink papular nodules that had been present for 3 months. Past medical history included cervical dysplasia with hysterectomy at the age of 29, hypothyroidism, a 10 mm tubular adenoma of colon and diagnosis of Lynch syndrome (2016), and SCC of the vulva (February 2016). Family cancer history included paternal thyroid cancer at age 52 and lymphoma at age 57 and maternal, endometrial pre-cancer at the age of 45.
Physical Examination: Oval-shaped, 6 mm, smooth, pink, papular nodules (R flank, R inframammary, R axilla) with scaling collarette at the periphery (on initial visit 2016)
Laboratory Data: CBC – normal, CMP – normal, CT of chest, abdomen, and pelvis –normal with no evidence of lymphomatous process, peripheral blood smear and flow cytometry - normal, mammogram normal, genetic mutation testing - positive for CHEK2 & MSH6.
Histopathology: Four of the five shave biopsies (over period of 3 months) revealed atypical, intravascular CD30+ T-cell lymphocytes (CD3+, CD5-).
Clinical Course: The patient developed 1-2 new lesions every 2 – 3 months that resolved with topical clobetasol cream. Newer lesions in 2017 which were biopsied: (a) smooth pink plaque on mid abdomen measuring 1.5 cm and (b) smooth red papule on left temple measuring 0.3 cm.
Diagnosis: Lymphomatoid papulosis variant with atypical intravascular CD30 positive lymphocytes – Type E Lymphomatoid Papulosis
Points of Emphasis: Lymphomatoid papulosis is a rare, recurrent and self-healing papulonodular disorder that belongs to the spectrum of primary cutaneous CD30+ lymphoproliferative disorders. The course of skin disease is chronic, often lasting years to decades. Clinically, it is characterized by grouped or generalized self-healing papulo-nodular lesions which wax and wane over weeks to months. Lesions may ulcerate, heal with scaling and crust, and in some instances, scar. Onset can happen from early childhood to middle age, however Caucasians are affected more frequently. Lymphomatoid papulosis is subclassified into 5 types (A, B, C, D, and, E) based on histologic findings. Type E is characterized clinically by papules that rapidly ulcerate. Histologically, Type E shows an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and usually CD8. The prognosis remains excellent with no extracutaneous spread or disease-related deaths. The histologic findings in this case fit most closely with angioinvasive lymphomatoid papulosis (Type E). The genetic mutations which were found in the patient also infer an increased risk of other cancers. CHEK2 mutations been shown to increase risk for breast, colorectal, and prostate cancer. MSH6 mutations are associated with Lynch syndrome, leukemia and lymphoma, and neurofibromatosis.
References: 1. Hughey, MD. Lauren C., Practical Management of CD30+ Lymphoproliferative Disorders. Dermatol Clinics: Cutan Lymphoma. October 2015; 819-8332. Wieser, MD. Iris, Oh, MD. Chee Won, Talpur, MD. Rakhshandra, Duvic, MD. Madeleine. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients J Am Acad Dermatol January 2016 Volume 74 Number 1; 59-67
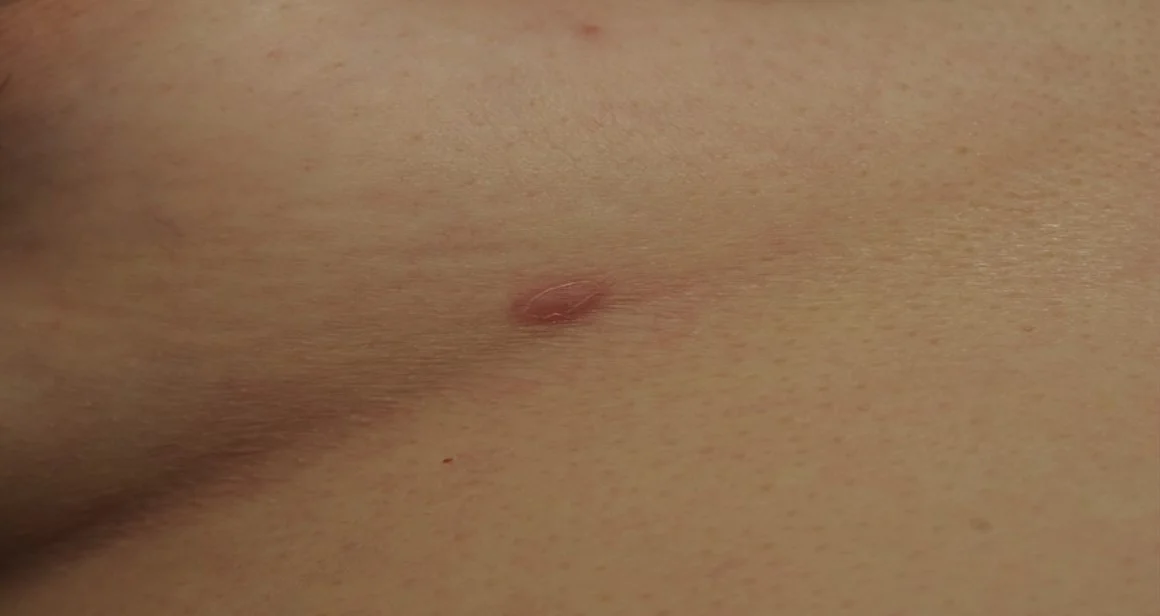
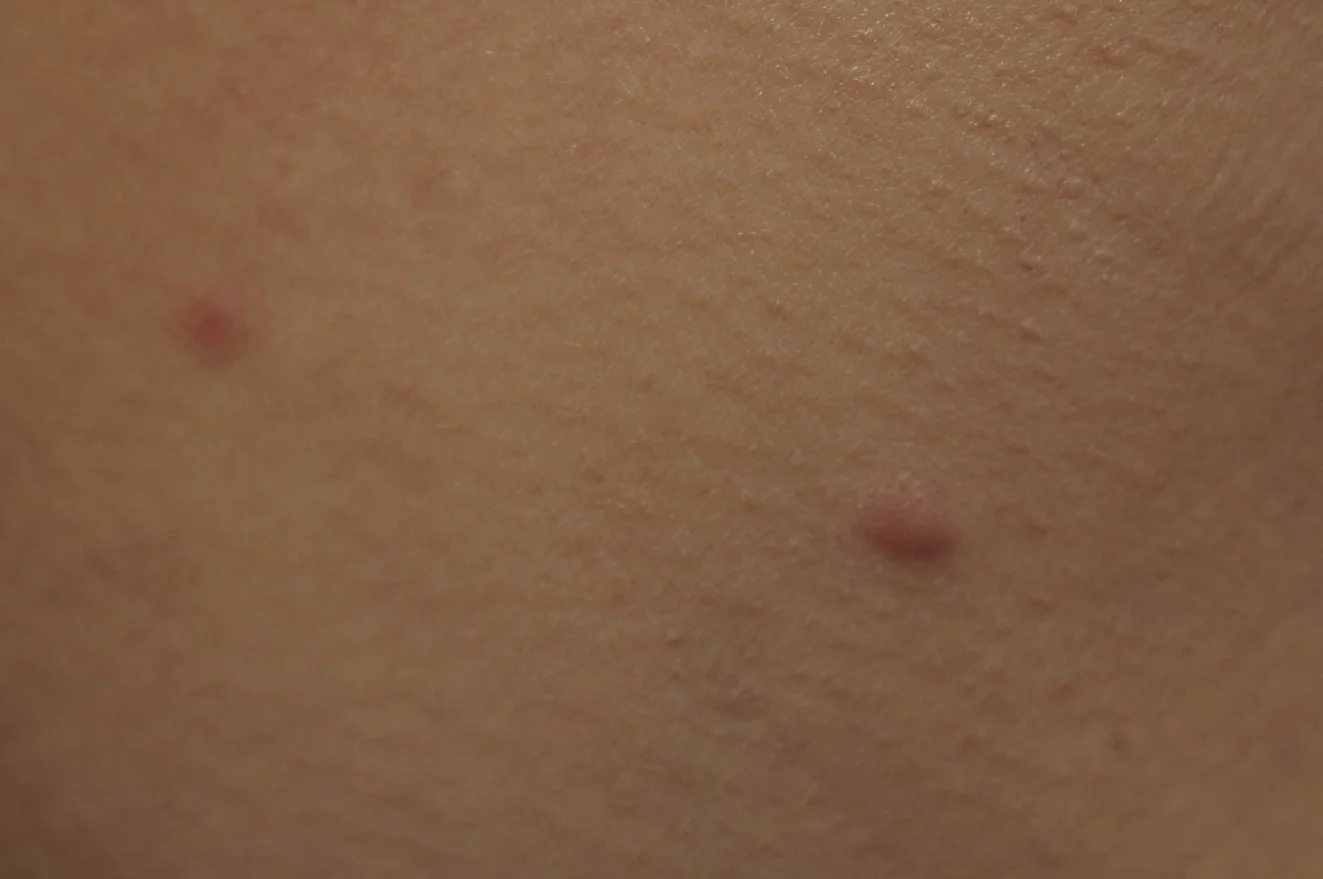










Case 22: The lesser known CD8+ Cutaneous T-Cell Lymphomas: What are they and why should we know about them?
Caregivers: Dipti Anand, M.D., Aditya Sood, UG-3, Atlanta and Athens, GA
History (Patient 1): A 65-year-old Caucasian female presented with a relatively acute onset of tender, erythematous subcutaneous nodules involving her right upper arm. She also reported constitutional symptoms including fever, myalgia and night sweats.
Physical Exam (Patient 1): Inflamed, erythematous subcutaneous nodules which were tender on palpation were noted on the right upper arm.
Histopathology (Patient 1): Initial biopsy showed marked fat necrosis with associated suppurative inflammation, worrisome for an infectious process. Special stains for infectious organisms (PAS, Gram, GMS, AFB & Fite), and tissue cultures were negative. A repeat biopsy showed a dense nodular & diffuse lobulocentric infiltrate in the pattern of lobular panniculitis. The infiltrate was composed of large pleomorphic lymphocytes with irregular nuclear contours & inconspicuous nucleoli. The tumor cells were arranged in small clusters & solitary units around individual adipocytes giving a “rimming” pattern. The overlying epidermis and dermis showed minimal change. The atypical cells were CD3, CD5, CD7, CD8, granzyme B, TIA-1, & BF1 + and negative for CD4, CD56, gamma M1 & CD30.
History (Patient 2): A 51-year-old Asian male presented with acute & rapidly progressive eruption of multiple crusted, hemorrhagic, ulcerated plaques & nodules, in a generalized distribution, involving his trunk & extremities.
Physical Exam (Patient 2): A papulonecrotic lesion with ulceration & hemorrhagic crust was noted on the right forearm. The clinical differential diagnoses included fixed drug eruption, PLEVA and infection.
Histopathology (Patient 2): Punch biopsy of the lesion from the right forearm showed an ulcerated epidermis extensively infiltrated by medium-sized, enlarged, hyperchromatic, atypical lymphocytes, also infiltrating the pilosebaceous units & eccrine glands. There was accompanying extensive hemorrhage. The neoplastic infiltrate was composed of CD3+ CD5+ CD8+ T-cells with rare scattered CD20+ B-cells. The neoplastic cells were also positive for CD2, CD56, BF1, TIA-1 & granzyme B and negative for gamma M1, CD4 and CD30.
Diagnosis: Patient 1: Subcutaneous panniculitis-like variant of cutaneous T-cell lymphoma. Patient 2: Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (generalized pagetoid reticulosis).
Points of Emphasis: Both “subcutaneous panniculitis-like variant of cutaneous T-cell lymphoma (SPLTCL)”and “primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (PCAECTCL) are rare but distinct types of CTCLs included in the “WHO Classification of Tumors of Haematopoietic & Lymphoid Tissues.” SPLTCL presents MC in middle aged/elderly females as a panniculitis with solitary or multiple, tender, inflamed, subcutaneous nodules, which may ulcerate, mimicking erythema nodosum, nodular vasculitis and lupus profundus. The lesions mostly involve the lower extremities, but arms, trunk, & face can be affected. Constitutional symptoms including fever, weight loss, myalgias & anemia are seen in ~50% patients. The histology shows a lobular panniculitis with “fat rimming” by neoplastic T-cells, which have an αβ cytotoxic CD8+ T-cell phenotype, also positive for CD3 and granzyme-B. The histologic differential diagnoses include Cutaneous γδ T-cell lymphoma (which lack the αβ gene rearrangement) & extranodal NK/T-cell lymphoma, nasal type (which is CD2, CD56 and EBV+). A history of autoimmune disorders, particularly LE, is present in ~20% cases. This lymphoma is a morphologic continuum of lupus panniculitis/profundus & atypical lymphocytic lobular panniculitis. Overall, it is an indolent lymphoma with good prognosis, and a 5-year overall survival of >80%. However, development of hemophagocytic syndrome in ~20% cases may portend a worse outcome. PCAECTCL (generalized pagetoid reticulosis/Ketron Goodman disease), is an extremely aggressive CD8+ T-cell lymphoma, presenting with a rapidly progressive, relentless, disseminated course of ulcerating plaques & tumor nodules. The lymphoma presents with widespread visceral metastases to uncommon sites including the testis, lungs, spleen, and central nervous system (without lymphadenopathy) and has a high mortality, with mean survival time of 12 months. Histology shows a mycosis fungoides-like epidermotropic T-cell infiltrate with ulceration & hemorrhage. The tumor cells are CD8+ and CD30 negative helping it to distinguish from lymphomatoid papulosis. In summary, it is important to be aware of these lesser known variants of cutaneous T-cell lymphomas which, unlike MF, can present as a panniculitis (SPLTCL) or with a rapidly progressive, fulminant course (PCAECTCL). Appropriate immunophenotypic work-up is needed to differentiate these tumors from others closely mimicking T-cell lymphomas.
References: 1. Slater, D.N., The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol, 2005. 153(5): p. 874-80. 2. Willemze, R., et al., Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood, 2008. 111(2): p. 838-45. 3. Mielke, V., et al., Localized and disseminated pagetoid reticulosis. Diagnostic immunophenotypical findings. Arch Dermatol, 1989. 125(3): p. 402-6.










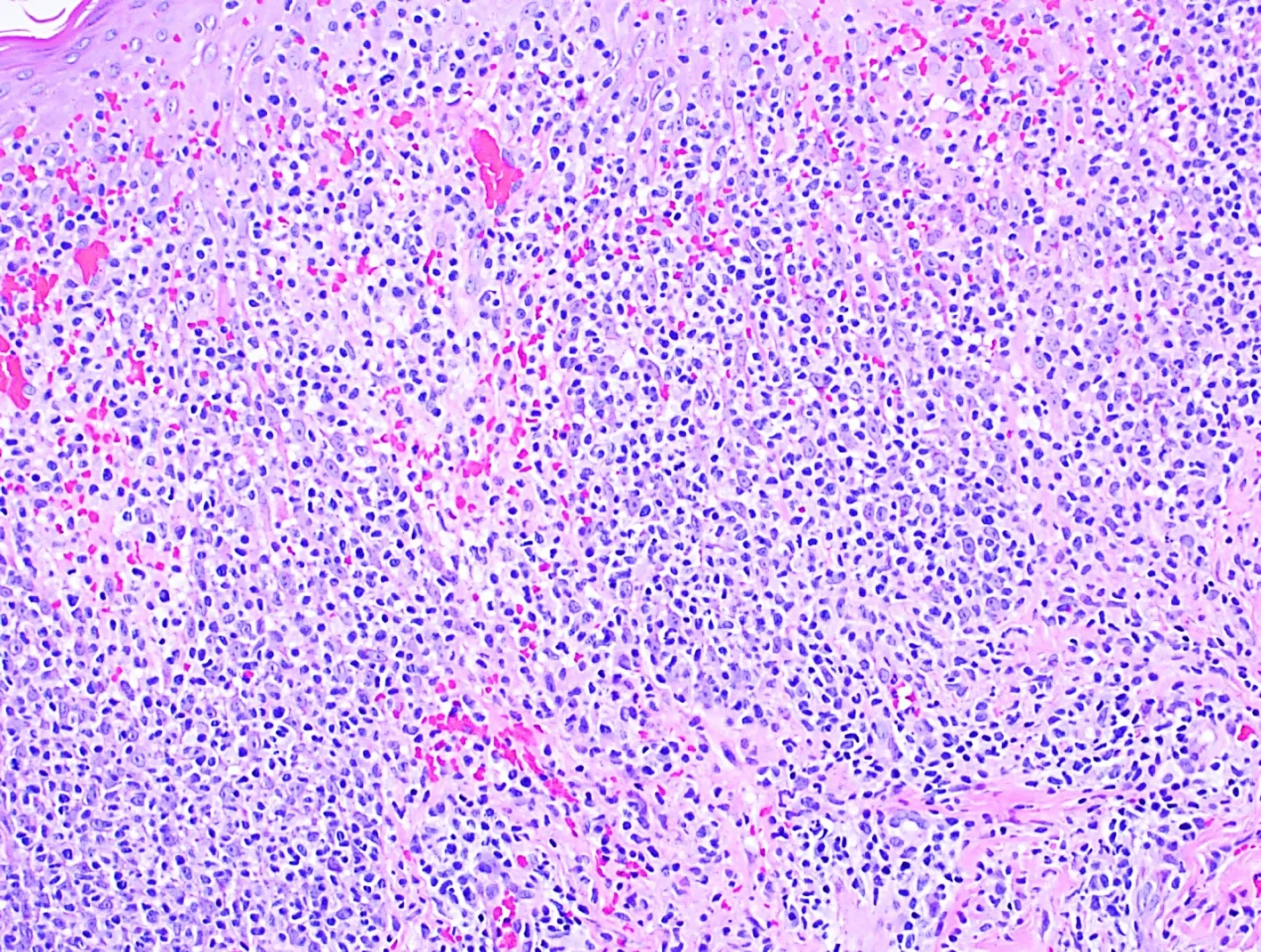




Case 23: Henoch-Schonlein Purpura
Caregivers: Rachel Evers-Meltzer, Lily Adelzadeh M.D., Azeen Sadeghian M.D., Lacey Sullivan M.D., Erin Boh, M.D., Ph.D., New Orleans, LA
History: A 70-year-old male with hypertension, COPD, CHF, and coronary artery disease was referred for management of leukocytoclastic vasculitis of unknown etiology. The patient developed petechiae and then palpable purpura on the distal lower extremities two years prior, which was diagnosed as biopsy-confirmed leukocytoclastic vasculitis. Clopidogrel was withheld for 3 months with no improvement. He failed therapy with colchicine, dapsone, and hydroxychloroquine. His vasculitis continued to progress despite continuous oral prednisone taken from the time of diagnosis to presentation, at which time the patient complained of steroid-related weight gain, muscle weakness, and knee and hip pain in addition to progressive ulcerated palpable purpuric lesions on the lower extremities, buttocks, and abdomen. Five weeks prior to his appointment, he was started on methotrexate but he was noncompliant due to perceived lack of benefit.
Physical Examination: General appearance: Cushingoid body habitus, central adiposity moonlike facies, dorsocervical fat pad present; Skin: Fine telangiectasias overlying the nose and cheeks; extensive symmetric palpable purpura densely distributed from ankle to knee radiating to thighs, buttocks, and lower abdomen; scattered symmetric ulcerated purpuric plaques with overlying eschar from ankles to lower abdomen; solitary 9mm necrotic ulceration on each flank
Laboratory Data: CBC: WBC 4.8, hemoglobin 11.3/hematocrit 34.8 (low); Urinalysis: WNL; ANA, cryoglobulins, c-ANCA, p-ANCA, RF negative; HIV, Hepatitis panel, RPR negative; Complement, total: >60 High; C3 and C4 were WNL.
Histopathology: Patient had two sets of punch biopsies: 2 years prior to this visit, a punch biopsy of the left flank was obtained, revealing neutrophilic infiltrate of vascular walls accompanied by nuclear debris, fibrin deposit in walls, and red cell extravasation. Findings most compatible with leukocytoclastic vasculitis. Punch biopsies from the left medial calf and abdomen were consistent with vasculitis. Direct immunofluorescence of both samples revealed IgA, IgM, C3, and fibrinogen in a vascular distribution, with mild vascular IgG deposition. Vasculitis with IgA deposition is compatible with Henoch-Schönlein Purpura.
Clinical Course: The patient was resumed on oral methotrexate 10mg/week, his oral prednisone was tapered to 10mg/day. After one month, the patient noticed some improvement. The methotrexate was then increased to 15mg/week with the goal of further decreasing the prednisone. The patient is also being monitored by nephrology, he has an upcoming renal scan and 24 hour urine collection.
Diagnosis: Henoch-Schönlein Purpura/IgA Vasculitis
Points of Emphasis: IgA vasculitis, also referred to as Henoch-Schönlein Purpura (HSP), is an immune complex small-vessel vasculitis primarily affecting the skin, gastrointestinal tract, joints, and kidneys. It is characterized by IgA immune deposits around small vessels, and on histology it is indistinguishable from leukocytoclastic vasculitis. The classic presentation includes lower-limb-predominant purpura, abdominal pain, and joint pain following a bacterial or viral infection. IgA vasculitis is the most common systemic vasculitis of childhood but is rare in adults. The adult form tends to be more severe than the self-limiting form typical of the pediatric population, with up to 20% of adults experiencing a relapsing form and up to 11% experiencing end-stage renal disease as a result of IgA vasculitis. Because the vast majority of cases of HSP are acute and self-limiting, treatment is generally symptomatic. Prednisone is considered standard therapy for single episodes of cutaneous LCV that are severe, but is generally not recommended as monotherapy for long-term therapy of persistent disease. Persistent or refractory cases may respond to azathioprine, mycophenolate mofetil, or cyclosporine. IVIG, plasmapheresis, infliximab, and rituximab have been reported to be beneficial in severe, refractory cutaneous LCV.
References: 1. Audemard-verger A, Pillebout E, Guillevin L, Thervet E, Terrier B. IgA vasculitis (Henoch-Shönlein purpura) in adults: Diagnostic and therapeutic aspects. Autoimmun Rev. 2015;14(7):579-85. 2. Chen KR, Carlson JA. Clinical approach to cutaneous vasculitis. Am J Clin Dermatol. 2008;9(2):71-92. 3. Micheletti RG, Werth VP. Small vessel vasculitis of the skin. Rheum Dis Clin North Am. 2015;41(1):21-32, vii.





Case 24: Acute Generalized Exanthematous Pustulosis Secondary to Intravenous Immunoglobulins
Caregivers: Valerie Shiu, M.D., Adaobi Nwaneshiudu Obasi, M.D., Ph.D., Sandra Osswald, M.D.; San Antonio, TX
History: A 36-year-old African American female with a history of mucous membrane pemphigoid was admitted to our facility for a worsening rash. The rash first appeared as itchy bumps in her inguinal folds and then spread to involve her trunk, upper and lower extremities, face, and axilla. She stated this rash was much different from her typical pemphigoid flares which involved her eyes, lips and airway. She endorsed skin burning, aching, and pruritus, unrelieved by topical steroids. She received intravenous immunoglobulin monthly for management of her mucous membrane pemphigoid with the last dose being 1 week prior to onset of this rash. About 1 month prior to this rash, she was started on diltiazem for management of hypertension. There were no other changes in her medication regimen.
Physical Examination: The patient was in moderate distress, febrile, uncomfortably lying in bed, and tearful during the exam. A full body skin exam revealed numerous clustered pustules on an erythematous base clustered to her bilateral axilla, groin, and pannus extending onto her trunk and extremities. She also had 2 large denuded plaques to her bilateral posterior thighs secondary to sheets of coalesced pustules.
Laboratory Data: A complete metabolic panel was within normal range without evidence of renal or liver involvement. A complete blood count was within normal without evidence of leukocytosis. Blood cultures were negative growth to date.
Histopathology: 3 shave biopsies were done. The fresh frozen specimen showed scattered and discrete subcorneal neutrophilic pustules, which was also demonstrated on H&E. DIF was positive for linear IgG and C3 along the basement membrane, consistent with her known diagnosis of mucous membrane pemphigoid.
Clinical Course: The patient’s IVIG and diltiazem were discontinued, and she was started on high dose corticosteroids IV. She was then transitioned to oral steroids and is undergoing a slow taper without flare of her disease.
Diagnosis: Acute generalized exanthematous pustulosis (AGEP)
Points of Emphasis: AGEP is a rare and serious reaction pattern characterized by an acute eruptive pustular dermatosis usually secondary to medication or infection. More than 90% of cases are medication related, with the most common being beta-lactam antibiotics. AGEP typically presents with high fever and numerous, small, non-follicular based pustules within a large area of erythema. Typical histologic findings are superficial, subcorneal pustules with a predominance of neutrophils and some eosinophils. Treatment involves withdrawal of the offending agent as well as topical corticosteroids and antipyretics.
References: 1. Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): A review and update. J Am Acad Dermatol. 2015 Nov;73(5):843-8. 2. Stern RS. Clinical practice. Exanthematous drug eruptions. N Engl J Med. 2012 Jun 28;366(26):2492-501.


Case 25: Granulomatous Folliculitis as a Wolf's Isotopic Response
Caregivers: Laura M. Corsini, M.D., H McDonald, H Siddiqui, C Kowalewski; San Antonio, TX
History: A 46-year-old man with a PMH of chronic lymphocytic leukemia treated with chemotherapy two years prior and now in remission presented to the ED with a one week history of a pruritic, erythematous rash first noticed on the forehead and neck. The rash subsequently spread to the face, abdomen and back. One month prior, the patient was admitted to the hospital for sepsis secondary to primary varicella zoster virus, confirmed by biopsy and DFA. The patient was treated with IV acyclovir followed by a course of PO valacyclovir, and complete resolution was observed in his clinic visit following discharge. The patient then presented two weeks later with the new rash as mentioned above.
Physical Examination: Rash consisted of diffusely distributed juicy, pink to red-brown papules located on the head, neck, and trunk – with all lesions located in the same sites as the initial lesions observed during the patient’s prior episode of varicella. The rash was also associated with bulky lymphadenopathy in the cervical and axillary chains.
Laboratory Data: Fungal serologies, Cryptococcus serum antigen, Coccidioides serology, RPR – all negative
Histopathology: Two punch biopsies were obtained and demonstrated a lymphohistiocytic and granulomatous reaction centered around hair follicles and adnexal structures. GMS and Fite stains were negative for fungal and mycobacterial organisms. CD3, CD20, and CD5 immunostains were consistent with a reactive process with strong CD3 and CD5 expression.
Clinical Course: The patient’s secondary rash was associated with mild pruritus, which was treated with daily cetirizine 10mg and triamcinolone 0.025% cream as needed. There was no evidence of infection or malignancy, and no further therapy was necessary. His lymphadenopathy was not associated with the rash, and this was further worked up by hematology. Unfortunately, recurrence of his CLL was discovered on lymph node biopsy and he is currently undergoing treatment.
Diagnosis: Granulomatous folliculitis as a Wolf’s isotopic response
Points of Emphasis: Wolf’s isotopic response (WIR) refers to the development of a distinctive skin disease at the site of another already healed and unrelated disease. The time period between the first and second pathologic process is variable, ranging from days to years. The most common initial disease is herpes zoster; however, WIR has also been described following herpes simplex and varicella. Once the initial skin disease has healed, a variety of dermatologic conditions may develop on the same site3. The most common isotopic responses are granulomatous reactions, including granuloma annulare, granulomatous vasculitis, sarcoidal granuloma and granulomatous folliculitis, as well as lichenoid reactions, cutaneous infections and malignancies. The exact mechanism of WIR is unknown. One theory is that the initial viral infection destroys local nerve fibers, which may alter the immune system of the affected skin. This would lead to hyperreactivity (leading to granulomatous or lichenoid dermatitis), local immunosuppression (tumor infiltrations or infectious diseases), or abnormal angiogenesis (vascular tumors). Lastly, viral particles may remain in the tissue and cause a delayed-type hypersensitivity reaction. Our case underscores the importance of performing a thorough physical exam and recording of the distribution of the disease when examining someone with herpes zoster, herpes simplex or varicella zoster. We also recommend a low threshold for performing an additional skin biopsy on any new presentation of a rash, particularly given that WIR can be the presenting sign of malignancy.
References: 1. Generalized granuloma annulare after varicella infection: Wolf isotopic response? Abbas O, Kurban M. J Am Acad Dermatol. 2014; 71:80-2. 2. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Lora V, Cota C, Kanitakis J. Dermatol Online J. 2014;20 3. Wolf's isotopic response: a series of 9 cases. Jaka-Moreno A, López-Pestaña A, López-Núñez M, Ormaechea-Pérez N, Vildosola-Esturo S, Tuneu-Valls A, Lobo-Morán C. Actas Dermosifiliogr. 2012;103:798-805.






Case 26: Acute Radiation Dermatitis with Koebnerized Psoriasis
Caregivers: Charles Perniciaro, M.D., Leonard Slazinski, M.D., Tampa and Sarasota, FL
History: A 52-year-old man with a long history of psoriasis (controlled with topical steroids) was diagnosed with adenocarcinoma of the prostate (Gleason 9) in 2003, and treated with radical prostatectomy. In 2013, because of a rising PSA, the patient was treated with leuprolide acetate, and later enzalutamide. In 2017 (at age 66), the man developed left hip pain and imaging disclosed bony metastasis. The patient was treated with localized radiotherapy to the left hip in May 2017 as well as taxotere and denosumab. He developed acute radiation dermatitis in the radiation field as well as a flare of his psoriasis, both in the radiation field and across the entire trunk and lower extremities.
Physical Examination: Mostly guttate papulosquamous lesions of psoriasis scattered across the trunk and lower extremities, with confluence into larger plaques in the radiation field on the buttocks.
Laboratory Data: PSA 28.7 ng/mL
Histopathology: Epidermal acanthosis with several necrotic keratinocytes and Civatte bodies, and focal spongiosis. Mitotic figures were easily identified. Focal areas with absence of the granular cell layer, and a parakeratotic crust containing a few neutrophils (Munro microabscess).
Clinical Course: At the patient’s request, the psoriasis was treated with topical triamcinolone, with gradual worsening over one month. Radiotherapy has been completed.
Diagnosis: Acute radiation dermatitis with koebnerized psoriasis
Points of Emphasis: The psoriasis was somewhat “masked” in the biopsy of acute radiation dermatitis. The Koebner response is the isomorphic phenemenon of psoriasis, whose description is attributed to Heinrich Koebner, a Jewish-German dermatologist in the 19th century.
References: 1. Ben-Yosef R, Soyfer V, Vexler A. Radiation therapy in cancer patients with psoriasis. The fractionated daily dose and the Koebner phenomenon. Radiother Oncol. 2005;74:21. 2. Charalambous H, Bloomfield D. Psoriasis and radiotherapy: exacerbation of psoriasis following radiotherapy for carcinoma of the breast (the Koebner phenomenon). Clin Oncol (R Coll Radiol). 2000;12:192-3.

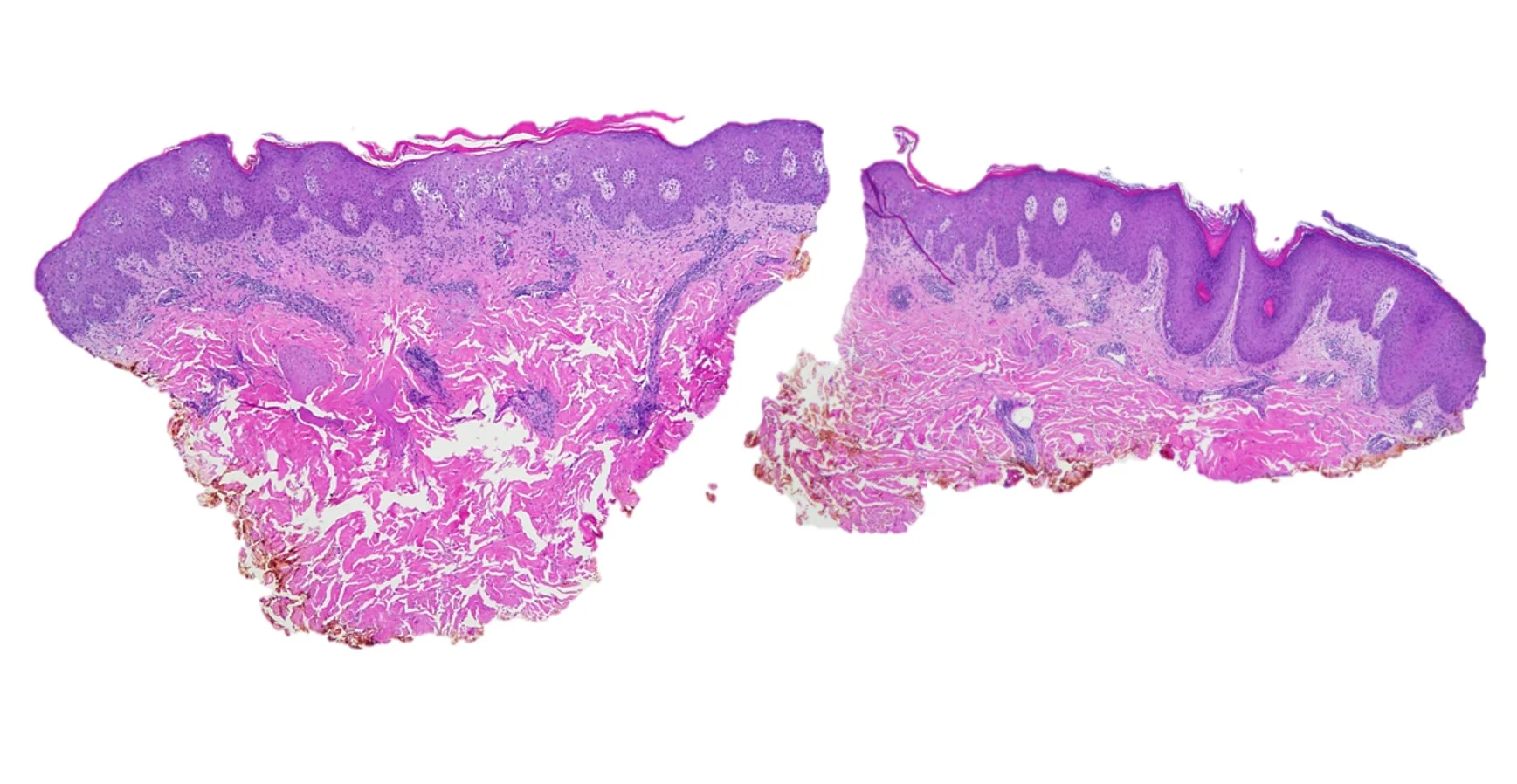



Case 27: Unmasking of Lupus Erythematosus with Imiquimod Therapy
Caregivers: Michael James Davis, BMus, Dipti Anand, MD, Atlanta, GA
History: A 58 year old Caucasian female with past medical history of hypertension and gastroesophageal reflux disease was referred by her PCP for further evaluation and treatment of “precancerous lesions”. Two lesions described as scaly, erythematous plaques on her chest were biopsied and reported as “actinic keratoses”. She was subsequently placed on 3.75% Imiquimod topical cream regimen (a two 2 week treatment cycle separated by a 2-week no treatment period). A follow-up visit after completion of the therapy showed appropriate reaction localized to chest lesions. However, a week after drug discontinuation, she returned with increased erythema of her chest lesions, some becoming confluent, with new erythematous plaques spreading to her back and upper extremities. She denied constitutional symptoms, and was treated with oral fexofenadine and prednisolone and topical corticosteroids, with improvement of her lesions within a week. However, approximately 2 weeks later, her rash worsened.
Physical Examination: She presented with crusted red plaques, with scale and vesiculation, on her neck, face and upper extremities with expanding and confluent lesions on her chest and back.
Laboratory Data: Serology studies showed positive antinuclear (1:200; diffuse homogenous pattern), anti-Ro/SSA (>8; reference range 0.0-0.9), and anti-histone antibodies, consistent with drug-induced subacute cutaneous LE.
Histopathology: Punch biopsies performed from the posterior neck and left dorsal forearm, showed an interface (vacuolar) dermatitis, with epidermal atrophy, hyperkeratosis, basal cell hydropic degeneration, scattered necrotic keratinocytes, basement membrane thickening, and accompanying perivascular and perifollicular lymphocytic inflammation. The findings were consistent with a connective tissue disease such as lupus erythematosus.
Clinical Course: She was started on prednisone taper & hydroxychloroquine, with subsequent improvement of her rash. The rash improved and nearly cleared approximately 3 months following therapy. Her last visit in January 2017 showed mild facial and chest erythema, and she is currently being followed concomitantly by Rheumatology.
Diagnosis: Imiquimod-induced subacute cutaneous lupus erythematosus
Points of Emphasis: Drug-induced lupus accounts for up to 10% of lupus erythematosus cases. Subacute cutaneous lupus erythematosus (SCLE) is a distinct clinical variant of lupus erythematosus that typically presents as annular, often scaly, erythematous plaques in photodistributed areas. Subacute cutaneous lupus erythematosus has been reported in association with multiple systemic medications including docetaxel, terbinafine, leuprolide acetate, etanercept, and efalizumab1-5; however, the induction of SCLE by topical agents has not been widely reported. We report a case of local induction of lesions that clinically and histologically resembled SCLE in a 58-year-old woman following treatment with topical imiquimod. Imiquimod is a topical immunomodulator used to treat genital warts and cutaneous malignancies. It exerts its effects via induction of proinflammatory cytokines, IFN-α, through activation of toll-like receptor (TLR) 7 signaling, & this may have been an inciting factor in the development of SCLE-like lesions in this likely genetically predisposed patient. It is possible that a subset of predisposed patients have a more robust TLR-7 reaction to imiquimod, resulting in abundant IFN-a production. Other autoimmune diseases, such as pemphigus foliaceus and vitiligo, also have been reported to occur locally after the application of imiquimod, which suggests that a localized autoimmune reaction can be induced by activation of TLR-7. It is important for one to be aware of this “immune activating” adverse effect of topical imiquimod, so that it can be used with caution, especially, in patients with pre-existing or family history of autoimmune diseases.
References: 1. Maguiness SM, Farsani TT, Zedek DC, Berger TG. Imiquimod-induced subacute cutaneous lupus erythematosus-like changes. Cutis. 2015; 95(6): 349-351. 2. Hejazi EZ, Werth VP. Cutaneous Lupus Erythematosus: An Update on Pathogenesis, Diagnosis and Treatment. Am J Clin Dermatol. 2016; 17(2): 135-46.






Case 28: Palmoplantar Eruption on Nivolumab
Caregivers: Jennifer Bares M.D., Elise Martin, Cather McKay M.D., and Erin Boh M.D., Ph.D., New Orleans, LA
History: A 70-year-old man with stage 4 non-small-cell lung cancer previously on Opdivo (nivolumab) presented to our clinic with 3-4 months of hyperkeratotic plaques on his palms and soles. He was started on nivolumab for treatment of his lung cancer 6 months prior to presentation. Approximately 3 months after starting nivolumab, he developed tender, red pustules on his bilateral hands. The pustules subsequently improved but were followed by the development of hyperkeratotic plaques on the bilateral fingers, toes, and feet. Lesions continued to thicken over time and began to disrupt nail growth on all four extremities. He denied any history of similar lesions as well as any arthritis, conjunctivitis or uveitis. Nivolumab was discontinued 2 weeks prior to presentation.
Physical Examination: Physical exam revealed an older, cachectic appearing man with multiple thick, brown, hyperkeratotic plaques and nodules on the distal palmar and dorsal hands and plantar feet, right worse than left; a 2x2 cm brown hyperkeratotic plaque on the left distal anterior lower leg, and generalized xerosis of the lower extremities.
Laboratory Data: Complete blood count and comprehensive metabolic panel were reviewed from previous and found to be within normal limits. Superficial fungal culture of the lesion was positive for Rhizopus.
Histopathology: A 4mm punch biopsy taken from the right lower leg demonstrated patchy lichenoid dermatitis, with staggered mounds of parakeratosis and neutrophils in the stratum corneum. Some sections showed a band-like lymphohistiocytic dermal infiltrate with associated basilar dyskeratotic keratinocytes. Occasional plasma cells were noted. GMS, Treponemal, and Warthin-Starry stains were negative.
Clinical Course: The patient was given urea 40% cream to apply daily to lesions and was seen for follow-up at one month. At that time, he reported interval improvement in the plaques on his hands and feet and denied development of new lesions. He remained off all treatment for his metastatic lung cancer with a clear PET-CT.
Diagnosis: Palmoplantar eruption in a patient on nivolumab for non-small-cell lung cancer.
Diagnostic consideration: While the diagnosis remains unclear, considering the clinical course, an immune reaction to nivolumab is most likely. Fungal culture of the lesion was positive for Rhizopus, so concurrent fungal super-infection cannot be excluded. However, it was felt the positive culture was most likely due to the patient being colonized with Rhizopus and treatment.
Points of Emphasis: Nivolumab is a fully human IgG4 anti-programmed cell death protein 1 (PD-1) receptor antibody. The inhibition of PD-1 results in the activation of T-cells and the cell-mediated immune response to tumor cells. While this upregulated immune response leads to antitumor activity, it can also induce immune-related side effects with the skin being one of the most commonly affected organs. In one, rash was the second most common adverse effect occurring in 23% of patients, second only to fatigue. A recent literature review published in 2016 found that rash, pruritus and vitiligo were the most commonly reported adverse cutaneous events in that order. Clinically, the rashes were described as erythematous macules, papules and plaques, typically on the trunk and extremities. On histopathology, lichenoid reaction/interface dermatitis was most commonly observed. Another single institution review found that 49% of patients developed adverse cutaneous events. The most common lesions were lichenoid reactions (17%), described as multiple, erythematous scaly papules and plaques, typically on the chest or back. This review also found that the incidence of developing these lesions increased at a constant rate after initializing treatment.
References: 1. Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020-1030. 2. Belum VR, Benhuri B, Postow MA, et al. Characterization and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-253. Hwang, S.J., Carlos, G., Wakade, D. et al.Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: A single-institution cohort. J Am Acad Dermatol. 2016; 74: 455–461.e1






Case 29: Keratosis Lichenoides Chronica
Caregivers: Laura Corsini, M.D., Anisha Guda, B.S., San Antonio, TX
History: A 46-year-old male with history of biopsy-proven keratosis lichenoides chronica of bilateral elbows and knees since 2013, most recently seen for a new lesion on the penis. Last visit (1/2017) biopsy of the penile lesion was consistent with the patient’s previous two biopsies from 2013 and 2014, favoring keratosis lichenoides chronica. The patient had been treated in the past with topical clobetasol ointment and intralesional kenalog. The patient had seen partial response with both therapies, but the lesions primarily involving his elbows and knees never resolved. They are intermittently itchy and sometimes painful. The patient noted that the condition worsened in the heat and on areas that had been traumatized.
Physical Examination: Purple-red, well-defined hyperkeratotic papules and plaques with overlying scale primarily clustered to the bilateral elbows and knees, with few scattered to the posterior upper arms, posterior thighs, and dorsum of the left hand. Left penile shaft with brown hyperpigmented linear patch with few pink hyperkeratotic papules.
Laboratory Data: RPR and HIV non-reactive.
Histopathology: All three shave biopsies (from 2013, 2014, 2017) showed hyperkeratosis with patchy parakeratosis, keratotic plugs, irregular epidermal acanthosis, vacuolar alteration with lichenoid infiltrate and ectatic vessels.
Clinical Course: The patient had been treated in the past with topical and intralesional corticosteroids, with some benefit. Given his extensive psychiatric history (which was not yet well-controlled), oral retinoid therapy as the next line treatment was avoided. Instead, he was started on calcipotriene and clobetasol ointment, as well as a topical retinoid (tazarotene cream) to use on the lesions daily. When the patient’s work schedule permits, we will next consider phototherapy 2-3x/week if needed.
Diagnosis: Keratosis lichenoides chronica (KLC)
Points of Emphasis: KLC is a rare dermatological condition characterized by violaceous-red hyperkeratotic papules and plaques on the limbs, frequently in a symmetric distribution. Lesions are generally asymptomatic or mildly pruritic and may be in a linear or reticulate pattern. Over half of patients experience facial involvement with seborrheic dermatitis or rosacea-like lesions. Other manifestations, such as palmoplantar keratoderma, mucosal involvement, nail dystrophy, and ocular manifestations occur in 20-30% of patients. KLC frequently appears in adolescence or adulthood with a chronic and progressive course; however, rare cases of spontaneous resolution have been reported. Treatment is challenging as the condition is often refractory to available therapies. Topical medications are usually ineffective (corticosteroids, salicyclic acid, tar), though literature has supported the successful use of topical calcipotriol. Other systemic therapies such as oral corticosteroids, antimalarial agents, methotrexate, gold, dapsone, erythromycin, radiotherapy and cyclosporine have also proved to be generally ineffective. The best results have been reported with phototherapy (NBUVB and especially PUVA) and oral retinoids. More effective treatment options for KLC are being researched for the future, including potentially biologic therapy (such as etanercept).
References: 1. Adisen E, et al. Easy to diagnosis, difficult to treat: keratosis lichenoides chronica. Clin Exp Dermatol. 2009; 35: 47-50. 2. Böer A. Keratosis lichenoides chronica: proposal of a concept.AmJ Dermatopathol. 2006; 28(3): 260-275.

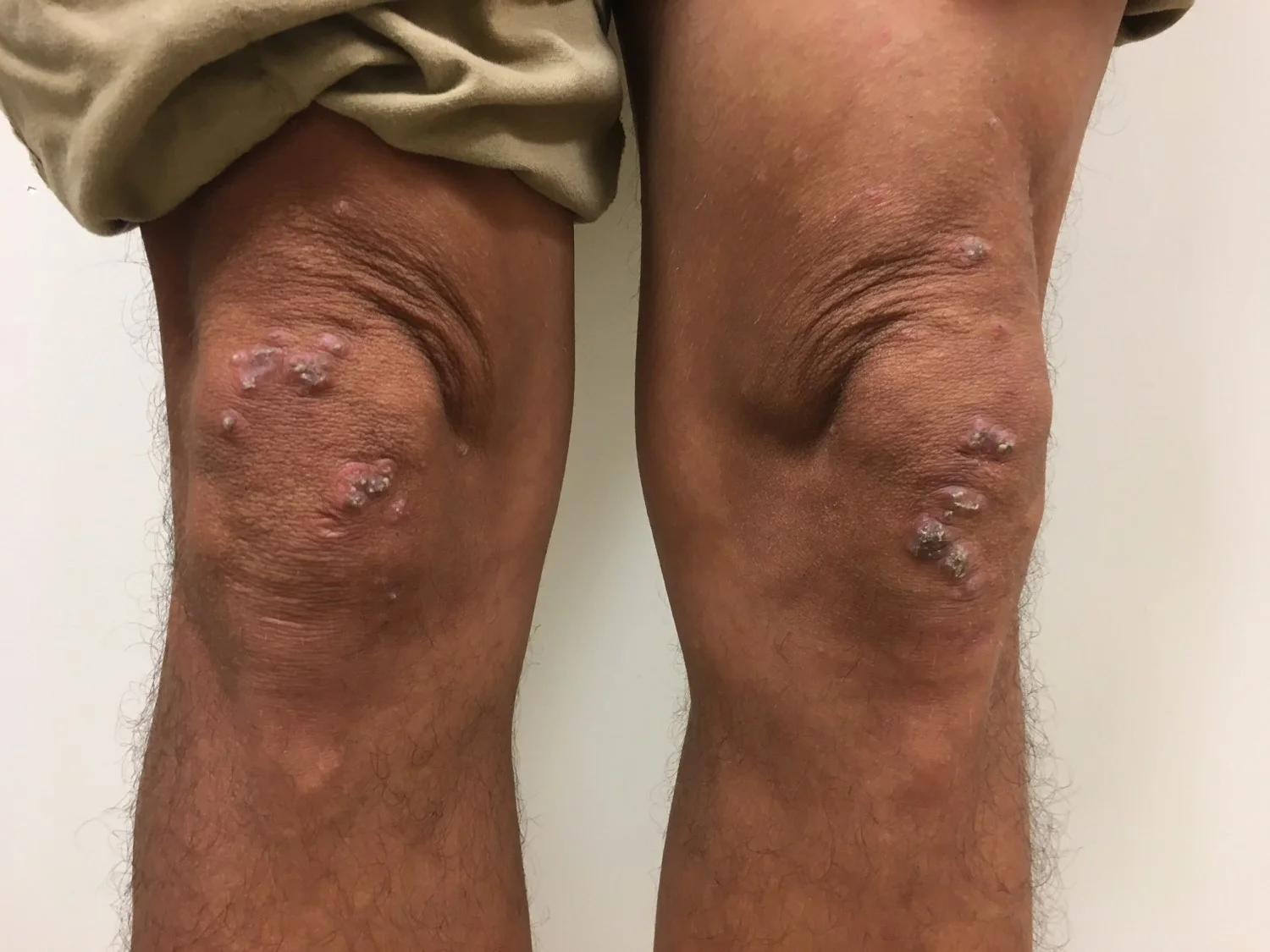



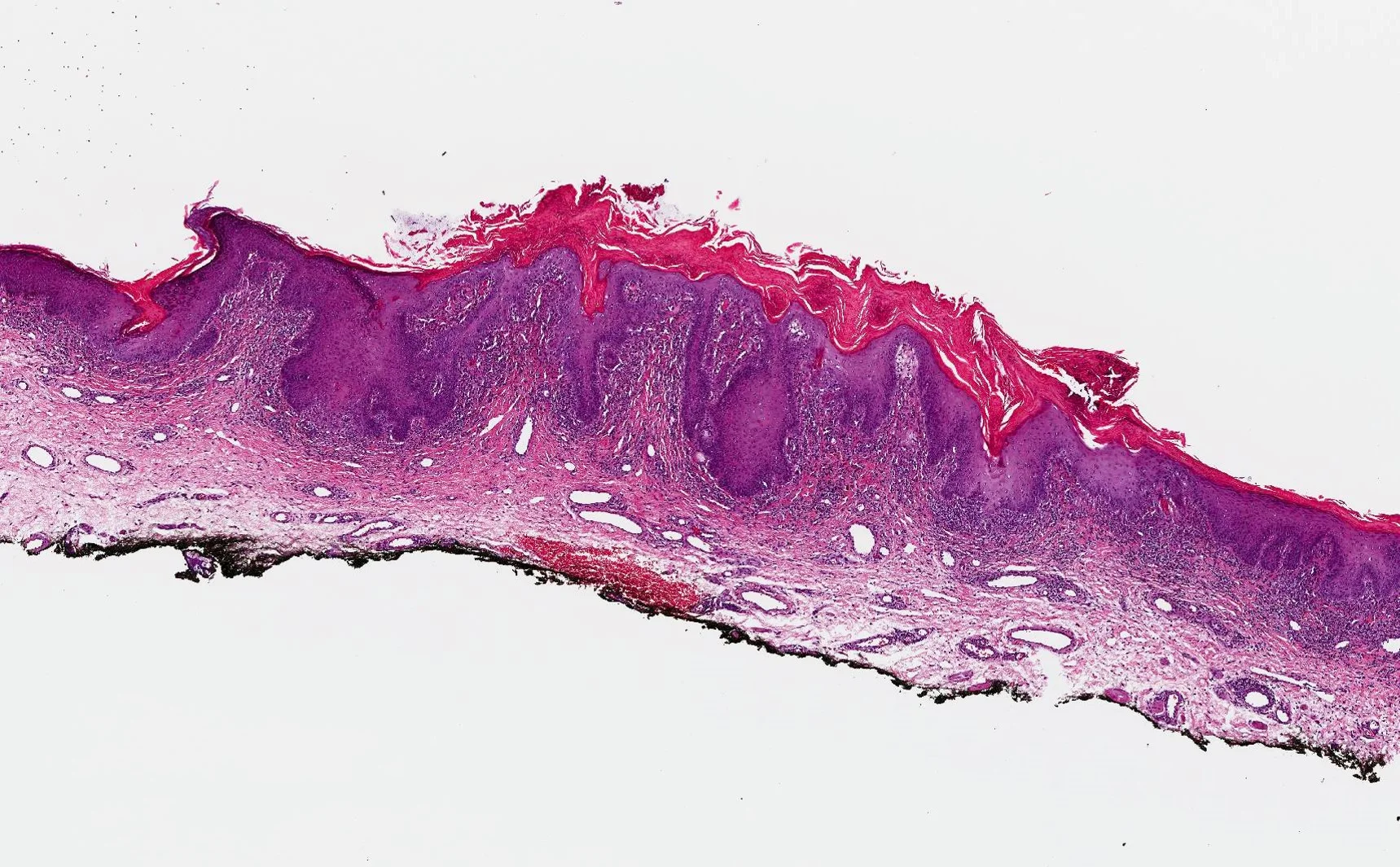

Case 30: Paraneoplastic Psoriasiform Necrolytic Erythema
Caregivers: Michael James Davis, BMus; Dipti Anand, M.D., Atlanta, GA
History: An 80-year-old Caucasian male presented with a crusted, scaly, erythematous eruption present in a periorificial and acral distribution involving the dorsal aspect of the hands, anterior cheek, chin, and extending onto the upper neck. The clinical differential diagnoses included eczematous contact dermatitis and acrodermatitis enteropathica.
Physical Examination: Erythematous, crusted, scaly plaques involving dorsal aspect of hands, anterior cheek, chin, and extending onto the upper neck.
Laboratory Data: Hepatitis C negative, zinc levels within normal limits, systemic work-up negative for underlying malignancy.
Histopathology: A punch biopsy from the right chin showed a psoriasiform dermatitis with confluent parakeratosis, hyperkeratosis, focal hypogranulosis, and neutrophilic exocytosis with formation of subcorneal microabscesses.
Clinical Course: The patient reported improvement of his rash following treatment with topical 0.1% triamcinolone & 0.05% fluocinonide ointments on his face and hands respectively. On further follow-up, approximately 1 year following the onset of his cutaneous eruption, he underwent lymph node biopsy of the groin and was diagnosed with Non-Hodgkin’s lymphoma.
Diagnosis: Paraneoplastic psoriasiform necrolytic erythema associated with non-Hodgkin’s lymphoma
Points of Emphasis: “Psoriasiform necrolytic erythemas” encompasses a group of cutaneous diseases of diverse metabolic origin that share overlapping clinical and histopathologic features. Included under this category are diseases such as acrodermatitis enteropathica, glucagonoma syndrome/necrolytic migratory erythema, necrolytic acral erythema, Hartnup disease, and pellagra. Clinically, patients present with a crusted erythematous, eczematous eruption, mimicking psoriasis or an eczematous dermatitis. Histology shows psoriasiform acanthosis with confluent parakeratosis, hyperkeratosis, mild spongiosis, hypogranulosis, and pallor of upper epidermal keratinocytes (“necrolysis”). The histology in certain cases, especially in late lesions, can be indistinguishable from psoriasis. An underlying nutritional and metabolic deficiency of trace elements like zinc & selenium, vitamins like niacin & biotin, and essential amino acid acids like tryptophan & glutamine, is the likely pathogenetic mechanism of the cutaneous eruption. This psoriasiform necrolytic eruption can also occur as a paraneoplastic phenomenon associated with or preceding the onset of a hematologic malignancy such as chronic myelogenous leukemia & Non-Hodgkin’s lymphoma. In summary, it is important for both clinicians and pathologists to be aware of the spectrum of “psoriasiform necrolytic dermatoses”. These groups of diseases can mimic the more common inflammatory cutaneous eruptions such as eczema and psoriasis, both clinically and histologically. They can be associated with an underlying nutritional or metabolic deficiency, but can also occur as a paraneoplastic manifestation of a systemic visceral or hematologic malignancy.
References: 1. Dasanu CA, Bauer F, Alexandrescu DT. Paraneoplastic psoriasiform dermatitis preceding the diagnosis of chronic myelogenous leukemia. Leuk Res. 2010; 34(12):e331-3. 2. Kovacs RK, Korom I, Dobozy A, Farkas G, Ormos J, Kemeny L. Necrolytic Migratory Erythema. J Cutan Pathol. 2006; 33(3): 242-5. 3. Stavropoulos PG, Papafragkaki DK, Avgerinou G, Papafragkakis H, Katsavou A, Katsambas AD. Necrolytic migratory erythema: a common cutaneous clue of uncommon syndromes. Cutis. 2013; 92(5): E1-4.






Case 31: Rowell's Syndrome
Caregivers: Jordan Maxwell Ward, B.S., Jaclyn N. Hess, M.D., Loretta S. Davis, M.D., Daniel J. Sheehan, M.D., Augusta, GA
History: A 39-year old African American female with a 15-year history of systemic lupus erythematosus (SLE), Sjogren’s syndrome, and Raynaud’s syndrome presented with a 6-day history of a worsening rash that began on her face and spread to the rest of her body. The rash was itchy and painful with extensive involvement of her lips and hard palate. Prior to this skin eruption, she had symptoms that were similar to her prior flares of SLE. Her rheumatologist had been trying to decrease her hydroxychloroquine and methotrexate doses and had halved her hydroxychloroquine dosage 6 months prior. The patient endorsed excess sun exposure 3 days prior to the eruption. Initially, she presented to her allergist who gave her an intramuscular injection of corticosteroid and started her on 60 mg of prednisone daily; however, for the following 3 days she continued to worsen. She denied any new, intermittent or continuous medications (specifically no NSAIDs). She also denied any fever blisters, sore throat or joint pain. A punch biopsy was performed on a targetoid-like lesion.
Physical exam: Diffuse necrotic vesicles and bullae on a dusky, erythematous base with atypical targetoid papules accompanied by central necrosis and surrounding erythema involving the face, conchal bowls, chest, arms, hands, abdomen, back, and legs. Diffuse erythema and purpura of the bilateral hands was noted as well as bilateral conjunctival injection with a thin line of crusted erosion along the lower eyelid margin bilaterally.
Histopathology: Basket-weave stratum corneum with underlying broad epidermal necrosis and vacuolar interface changes consistent with erythema multiforme. An area of frank clefting between the epidermis and dermis was noted.
Clinical Course: Serology showed positive anti-Ro, anti-dsDNA, anti-Sm antibodies, and a negative rheumatoid factor. The patient was initiated on IV methylprednisolone 1 gram daily for 3 days followed by prednisone 60 mg/day in addition to supportive care with IV fluids and pain control. She was stable at discharge with her symptoms progressively resolving.
Diagnosis: Rowell’s Syndrome.
Points of Emphasis: Rowell’s syndrome (RS) is a rare syndrome with diagnostic criteria which includes three major criteria (SLE, discoid lupus erythematosus, or subacute cutaneous lupus erythematosus; erythema multiforme-like lesions, with or without mucosal involvement; and an antinuclear Antibody (ANA) with a speckled pattern) and three minor criteria (chilblains, anti-Ro and anti-La antibodies, and a positive rheumatoid factor.). All three major criteria and at least one minor criterion are required for a diagnosis of RS. In this patient’s case, an ANA was not obtained since the patient had been diagnosed with SLE 15 years prior. However, since she is positive for anti-Ro and anti-Smith antibodies then, by definition, she has antibodies with a speckled pattern. In recent years, there have been debates regarding whether RS is a distinct diagnosis versus a coincidental overlap of 2 distinct diseases or even a variant of subacute cutaneous lupus. As more patients present with clinical and serologic findings consistent with RS, the diagnostic criteria should be re-evaluated. The authors propose using more specific antibody criteria, such as anti-Smith and anti-Ro (dsDNA) antibodies, rather than a speckled pattern in the development of new diagnostic criteria for RS. In patients with new onset targetoid-like lesions and a history of SLE or one of its variants, the diagnosis of Rowell’s syndrome is important to consider.
References: 1. Zeitouni NC, Funaro D, Cloutier RA, Gagné E, Claveau J. Redefining Rowell's syndrome. Br J Dermatol. 2000 Feb; 142(2):343-6. 2. Bhat RY, Varma C, Bhatt S, Balachandran C. Rowell syndrome. Indian Dermatology Online Journal. 2014;5(Suppl 1):S33-S35. 3. Kumar Y, Bhatia A, Minz RW. Antinuclear antibodies and their detection methods in diagnosis of connective tissue diseases: a journey revisited. Diagnostic Pathology. 2009;4:1.
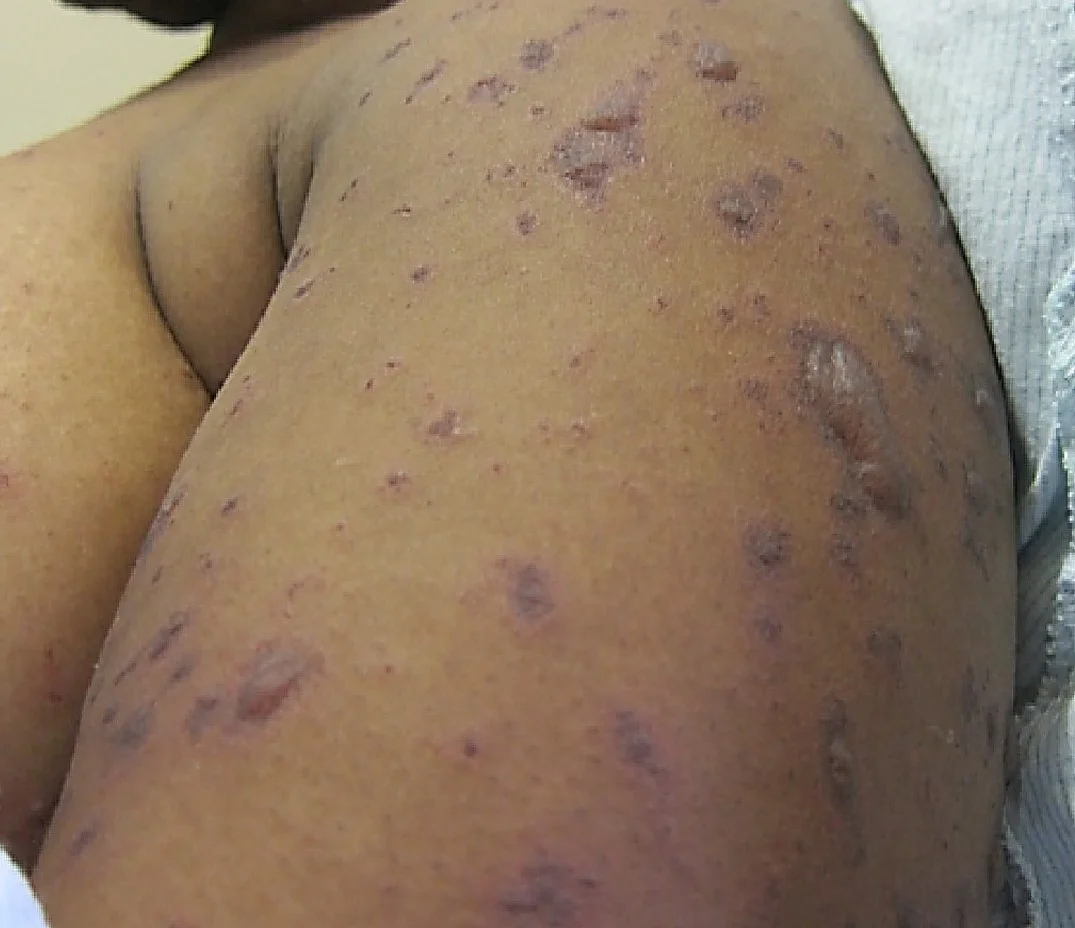





Case 32: H Syndrome
Caregivers: Sarah McGinty, M.D., Hoang Ho-Pham, MS-IV, Pamela Martin, M.D., New Orleans, LA
History: A 55-year-old female of Arab descent presented for evaluation of a progressive rash on the abdomen, back, and bilateral lower extremities, present for over 10 years. The rash was associated with pruritus and discoloration. She had been treated previously with topical steroids and antihistamines which provided some relief, but the rash persisted. Past medical history included anemia, deafness, hypothyroidism, atrial myxoma, congestive heart failure, and chronic hepatosplenomegaly. Of note, the patient had been seen by multiple doctors who all agreed her symptoms were likely a product of some genetic syndrome, but a formal diagnosis was never made.
Physical Examination: Coarse facies, short stature (60 in), abdomen and lower back with hyperpigmented, indurated plaques with reticulated borders; bilateral medial thighs with hyperpigmented indurated plaques, edema and hyperpigmentation extending to ankles/feet with sparing of knees; contracture and ulnar deviation of DIP joints on bilateral hands
Laboratory Data: Hgb 10.1g/dl [12.0-16.0g/dl], Hct 30.8% [37-47%], RF(-), Scl70(-), ANA(-)
Histopathology: Punch biopsy of right lateral thigh revealed epidermal acanthosis and hyperpigmentation. Dermal vessels with lymphoid cuffing and associated hemosiderin deposits. S100 positive, CD68 and CD163 weakly positive, CD1a negative histiocytic infiltrate of dermis with calcific hyperelastosis, and focal dermal mucin deposits. Some histiocytes exhibit intracytoplasmic lymphocytes/emperipolesis.
Clinical Course: Skin symptoms currently controlled with Triamcinolone 0.1% cream, Sarna sensitive lotion, Atarax 10 mg qhs, SLC29A3 genetic testing pending.
Diagnosis: Probable H syndrome
Points of Emphasis: H syndrome is a rare autosomal recessive genodermatosis with multisystem involvement seen most often in patients of Arab ancestry. It is characterized by specific clinical features including hyperpigmentation, hypertrichosis, hepatosplenomegaly, hearing loss, heart anomalies, hypogonadism, low height, hyperglycemia and hallux valgus/flexion contractures. Onset typically occurs in the first or second decade of life. The hallmark of H syndrome is the hyperpigmented, hypertrichotic, sclerodermatous plaques found on bilateral thighs. These changes are common in affected patients and can be considered pathognomonic. The second and third most common clinical findings include flexion contractures of the hands and feet and sensorineural hearing loss respectively. In addition to symptoms discussed above, it is reported that patients may also have lymphadenopathy, microcytic anemia, renal and skeletal abnormalities. Cutaneous histopathology reveals hyperpigmentation of the basal layer with acanthosis, marked fibrosis with histiocytic infiltrate and perivascular mononuclear infiltrate. The histiocytes are CD68 positive, S100 positive, CD1a negative, simulating Rosai-Dorfman disease. Histiocytes may also exhibit intracytoplasmic inflammatory cells/emperipolesis. Affected individuals demonstrate mutations in SLC29A3. This gene encodes human equilibrative nucleoside transporter 3 (hENT3) which is thought to transfer nucleosides from lysosomes to cytoplasm and across the inner mitochondrial membrane. Defects in hENT3 disrupt the homeostatic functions of lysosomes and mitochondria in affected patients producing the phenotype described above. Genetic analysis is considered confirmatory for this entity. However, Mohlo et al maintain that the diagnosis of H syndrome can be made given the specific pattern of hyperpigmentation and sclerodermatous skin changes on bilateral thighs in addition to systemic symptoms. Therapeutic trials with systemic corticosteroids and other immunosuppressive agents have resulted in partial or no response.
References: 1. Molho-Pessach V, Ramot Y, Camille F et al. H Syndrome: The first 79 patients. J Am Acad Dermatol 2014; 70 (1):80-88. 2. Molho-Pessach V, Agha Z, Aamar S et al. The H syndrome: a genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol 2008; 59(1): 79-85.








Case 33: Capillary Malformations with RasA1 Mutation
Caregivers: Emily Croce, RN-CPNP, Lucia Diaz, M.D., Austin, TX
History: An otherwise healthy Hispanic boy presented to pediatric dermatology for initial evaluation at 4 years of age. He was born with a red, vascular patch on his left thigh and scrotum that had grown with him. At age 3, he began to develop new red, vascular patches on his face, torso, and extremities. None of the areas were warm, tender, or ever had any associated swelling/pain. There was no history of headaches or other symptoms. There was no known family history of similar patches on the skin or other significant medical problems.
Physical Examination:Well-appearing 4-year-old male in no apparent distress. No dysmorphic features. Red, vascular patch extending from left lateral scrotum to left medial, proximal thigh. Scattered, red, vascular, 1-4 cm patches on left chin, bilateral upper & lower extremities, back, abdomen. No areas with increased warmth, thrill, bruit, swelling.
Laboratory Data: ARUP Vascular Malformations Panel, Sequencing and Deletion/Duplication, 15 Genes
- Pathogenic Variant, c.2011+1G>C, was detected in the RASA1 gene.
- A variant of unknown significance, c.893_894delinsAA; p.Cys298Ter was detected in the TEK/TIE2 gene.
These two variants have not previously been identified by the ARUP Laboratory, are not listed in gene-specific databases, and have not been reported in medical literature. The RASA1 gene has been implicated in capillary malformation-arteriovenous malformation (CM-AVM) syndrome and Parkes Weber Syndrome. The TEK/TIE2 gene has been implicated in inherited, multiple, cutaneous and mucosal, venous malformations.
MRI of brain and spine, with/without contrast:
- Negative MRI of cervical, thoracic, and lumbar spine.
- Small, curvilinear area of gliosis and intravascular enhancement between the left superior temporal gyrus and atrium of the left lateral ventricle of the brain. This is typically considered to be a developmental, venous anomaly, but can be seen associated with blue rubber bleb nevus syndrome.
Histopathology: 4 mm punch biopsy from an affected area on the left, lower back shows an increase in the number of small blood vessels in the papillary and superficial reticular dermis as well as around adnexal structures.
Clinical Course: The patient has continued to be healthy 1.5 years after initial presentation. He has developed additional, small, red, vascular patches on his skin. He was evaluated by a pediatric genetics clinic. His parents underwent genetic testing offered free of charge by ARUP to test for the same variant in the RASA1 gene. They were both negative for this variant, therefore patient’s mutation is de novo. They were not tested for the TEK/TIE2 gene variant. Our plan is to continue to follow up with him annually, and we will repeat MRI plus add gadolinium MRA, both with/without contrast, every 1-2 years.
Diagnosis: Multiple, acquired, cutaneous, capillary malformations in the setting of a de novo, pathogenic variant in the RASA1 gene and a variant of unknown significance in the TEK/TIE2 gene
Points of Emphasis: Patients who present with multiple, cutaneous and/or mucosal, capillary malformations are at risk of fast-flow arteriovenous and/or venous malformations, particularly of the brain and spine. Consideration should be given to brain/spine MRI/MRA and genetics consultation in this setting. Research is ongoing regarding which genetic variants are known to be pathogenic, and the exact monitoring protocol that these patients should undergo. If not de novo, pathogenic RASA1 and TEK/TIE2 variants are inherited in an autosomal dominant fashion and genetic counseling should be offered to the family of an affected individual.
References: 1. Frigerio, A, Stevenson, DA & Grimmer, JF. The genetics of vascular anomalies. Current Opinion in Otolaryngology & Head and Neck Surgery. 2012 Dec: 20(6):527-32. 2. Orme, CM et. al. Capillary malformation arteriovenous malformation syndrome: Review of the literature, proposed diagnostic criteria, and recommendations for management. Pediatric Dermatology. 2013 Jul-Aug;30(4):409-15





